El Variational Quantum Eigensolver (VQE)
Esta lección presenta el Variational Quantum Eigensolver, explica su importancia como algoritmo fundamental en la computación cuántica y examina sus fortalezas y debilidades. VQE por sí solo, sin métodos complementarios, probablemente no será suficiente para los cálculos cuánticos modernos a escala de utilidad. Sin embargo, es importante como método híbrido clásico-cuántico arquetípico y constituye un fundamento esencial sobre el cual se construyen muchos algoritmos más avanzados.
Este video ofrece una descripción general de VQE y los factores que afectan su eficiencia. El texto siguiente complementa esto con más detalles e implementa VQE con Qiskit.
1. ¿Qué es VQE?
El Variational Quantum Eigensolver es un algoritmo que utiliza conjuntamente la computación clásica y cuántica para resolver una tarea. Un cálculo VQE consta de cuatro componentes principales:
- Un operador: A menudo un hamiltoniano, al que llamamos , que describe una propiedad del sistema que se desea optimizar. En otras palabras, se busca el vector propio de este operador que corresponde al valor propio mínimo. A este vector propio lo llamamos frecuentemente el "estado fundamental".
- Un "ansatz" (una palabra alemana): Un circuito cuántico que prepara un estado cuántico que se aproxima al vector propio buscado. Estrictamente hablando, un ansatz es una familia de circuitos cuánticos, porque algunas de las puertas del ansatz están parametrizadas, es decir, se les pasa un parámetro que podemos variar. Esta familia de circuitos puede generar una familia de estados cuánticos que aproximan el estado fundamental.
- Un Estimator: Un medio para estimar el valor esperado del operador sobre el estado cuántico variacional actual. A veces, este valor esperado, la llamada función de costo, es nuestro objetivo principal. A veces nos interesa una función más compleja que, sin embargo, se compone de uno o más valores esperados.
- Un optimizador clásico: Un algoritmo que varía parámetros para minimizar la función de costo.
Examinemos cada uno de estos componentes con más detalle.
1.1 El operador (hamiltoniano)
En el centro de un problema VQE se encuentra un operador que describe un sistema de interés. Aquí asumimos que el valor propio más pequeño y el vector propio asociado de este operador son útiles para un propósito científico o práctico. Ejemplos de esto incluyen un hamiltoniano químico que describe una molécula, donde el valor propio más pequeño corresponde a la energía del estado fundamental de la molécula y el estado propio asociado describe la geometría o configuración electrónica de la molécula. O bien, el operador describe el costo de un proceso a optimizar, y los estados propios corresponden a rutas o estrategias. En algunos campos como la física, un "hamiltoniano" casi siempre se refiere a un operador que describe la energía de un sistema físico. Sin embargo, en la computación cuántica es común referirse a operadores de mecánica cuántica que describen un problema empresarial o logístico también como "hamiltoniano". Adoptamos esta convención aquí.

Mapear un problema físico o de optimización a qubits no es típicamente una tarea trivial, pero estos detalles no son el foco de este curso. Una discusión general sobre el mapeo de un problema a un operador cuántico se encuentra en Quantum computing in practice. Una mirada más detallada al mapeo de problemas químicos a operadores cuánticos se ofrece en Quantum Chemistry with VQE.
Para los fines de este curso, asumimos que la forma del hamiltoniano es conocida. Un hamiltoniano para una molécula simple de hidrógeno (bajo ciertas suposiciones de espacio activo y con el mapeador de Jordan-Wigner) es, por ejemplo:
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy
from qiskit.quantum_info import SparsePauliOp
hamiltonian = SparsePauliOp(
[
"IIII",
"IIIZ",
"IZII",
"IIZI",
"ZIII",
"IZIZ",
"IIZZ",
"ZIIZ",
"IZZI",
"ZZII",
"ZIZI",
"YYYY",
"XXYY",
"YYXX",
"XXXX",
],
coeffs=[
-0.09820182 + 0.0j,
-0.1740751 + 0.0j,
-0.1740751 + 0.0j,
0.2242933 + 0.0j,
0.2242933 + 0.0j,
0.16891402 + 0.0j,
0.1210099 + 0.0j,
0.16631441 + 0.0j,
0.16631441 + 0.0j,
0.1210099 + 0.0j,
0.17504456 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
],
)
Nótese que el hamiltoniano anterior contiene términos como ZZII e YYYY que no conmutan entre sí. Para evaluar ZZII, entre otras cosas, habría que medir el operador Pauli-Z en el qubit 3. Sin embargo, para evaluar YYYY, hay que medir el operador Pauli-Y en el mismo qubit 3. Entre el operador Y y el Z en el mismo qubit existe una relación de incertidumbre; ambos operadores no pueden medirse simultáneamente. Volveremos sobre este punto más adelante y a lo largo del curso.
El hamiltoniano anterior es un operador matricial de . Diagonalizar el operador para determinar su valor propio de energía más pequeño no es difícil.
import numpy as np
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues), "hartrees")
The ground state energy is -1.1459778447627311 hartrees
Los solucionadores de valores propios clásicos por fuerza bruta no pueden escalar para describir las energías o geometrías de sistemas atómicos muy grandes como fármacos o proteínas. VQE es uno de los primeros intentos de hacer que la computación cuántica sea útil para este problema.
En esta lección nos encontraremos con hamiltonianos significativamente más grandes que el anterior. Sin embargo, sería un desperdicio forzar los límites de lo que VQE puede hacer antes de que más adelante en este curso presentemos algunas de las herramientas más avanzadas que pueden complementar o reemplazar a VQE.
1.2 Ansatz
La palabra "ansatz" es alemana. El plural correcto en alemán es "Ansätze", aunque también se encuentran frecuentemente "ansatzes" o "ansatze". En el contexto de VQE, un ansatz es el circuito cuántico con el que se genera una función de onda de múltiples qubits que se aproxima lo mejor posible al estado fundamental del sistema estudiado y, por lo tanto, proporciona el valor esperado más pequeño del operador. Este circuito contiene parámetros variacionales (a menudo agrupados en el vector de parámetros ).

Se elige un conjunto inicial de valores de los parámetros variacionales. A la operación unitaria del ansatz en el circuito la llamamos . Por defecto, todos los qubits en las computadoras cuánticas de IBM® se inicializan en el estado . Cuando se ejecuta el circuito, el estado de los qubits es:
Si solo necesitáramos la energía más baja (en el lenguaje de los sistemas físicos), podríamos estimarla midiendo la energía muchas veces y tomando el valor más pequeño. Sin embargo, típicamente también queremos conocer la configuración que produce esta energía más baja o este valor propio mínimo. El siguiente paso es, por lo tanto, la estimación del valor esperado del hamiltoniano mediante mediciones cuánticas. Esto implica bastante. Cualitativamente, este proceso se puede entender considerando que la probabilidad de medir una energía (de nuevo en el lenguaje de los sistemas físicos) está relacionada con el valor esperado mediante la siguiente relación:
La probabilidad está además relacionada con el solapamiento entre el estado propio y el estado actual del sistema :
Mediante muchas mediciones de los operadores de Pauli que componen nuestro hamiltoniano, podemos estimar el valor esperado del hamiltoniano en el estado actual del sistema . El siguiente paso consiste en variar los parámetros y aproximarse aún más al estado fundamental del sistema con la energía más baja. Debido a los parámetros variacionales en el ansatz, a veces también se le denomina forma variacional.
Antes de abordar este proceso variacional, a menudo es útil partir de un "buen estado inicial". Si se conoce lo suficiente sobre el sistema, se puede hacer una mejor estimación inicial que . Por ejemplo, en aplicaciones químicas es común inicializar los qubits en el estado de Hartree-Fock. Este estado inicial sin parámetros variacionales se denomina estado de referencia. Al circuito para generar el estado de referencia lo llamamos . Siempre que sea importante distinguir el estado de referencia del resto del ansatz, utilice: De manera equivalente:
1.3 Estimator
Necesitamos una forma de estimar el valor esperado de nuestro hamiltoniano en un estado variacional determinado . Si pudiéramos medir directamente el operador completo , sería tan sencillo como realizar muchas (digamos ) mediciones y promediar los valores medidos:
El símbolo nos recuerda que este valor esperado solo sería exacto en el límite . Sin embargo, con miles de mediciones en un circuito, el error de muestreo del valor esperado es bastante pequeño. Para cálculos muy precisos, otros factores como el ruido juegan un papel importante.
No obstante, en general no es posible medir de una sola vez. puede contener múltiples operadores de Pauli X, Y y Z que no conmutan entre sí. Por lo tanto, el hamiltoniano debe dividirse en grupos de operadores que puedan medirse simultáneamente, y cada grupo debe estimarse por separado; luego se combinan los resultados para obtener un valor esperado. Abordaremos esto con más detalle en la siguiente lección, cuando discutamos la escalabilidad de los enfoques clásicos y cuánticos. Esta complejidad en la medición es una razón por la que necesitamos código altamente eficiente para este tipo de estimación. En esta lección y las siguientes, utilizamos para ello la primitiva Estimator del entorno de ejecución de Qiskit.
1.4 Optimizadores clásicos
Un optimizador clásico es un algoritmo clásico diseñado para encontrar valores extremos de una función objetivo (típicamente un mínimo). Busca en el espacio de parámetros posibles un conjunto que minimice una función de interés. En términos generales, estos algoritmos pueden clasificarse en métodos basados en gradientes, que utilizan información del gradiente, y métodos libres de gradientes, que funcionan como optimizadores de caja negra. La elección del optimizador clásico puede afectar significativamente el rendimiento de un algoritmo, especialmente en presencia de ruido en el hardware cuántico. Entre los optimizadores comunes en este ámbito se encuentran Adam, AMSGrad y SPSA, que han mostrado resultados prometedores en entornos ruidosos. Entre los optimizadores más tradicionales se encuentran COBYLA y SLSQP.
Un flujo de trabajo típico (como se muestra en la Sección 3.3) consiste en utilizar uno de estos algoritmos como método dentro de un minimizador como la función minimize de SciPy. Esta acepta como argumentos:
- Una función a minimizar. A menudo es el valor esperado de la energía. En general, estas se denominan "funciones de costo".
- Un conjunto de parámetros iniciales para la búsqueda. Frecuentemente denominados o .
- Argumentos, incluyendo los argumentos de la función de costo. En computación cuántica con Qiskit, estos incluyen el ansatz, el hamiltoniano y la primitiva Estimator, que se describe con más detalle en la siguiente subsección.
- Un método de minimización (
method). Este es el algoritmo específico que busca en el espacio de parámetros. Aquí se especificaría, por ejemplo, COBYLA o SLSQP. - Opciones (
options). Las opciones disponibles pueden variar según el método. Un ejemplo que prácticamente todos los métodos incluyen es el número máximo de iteraciones del optimizador antes de detenerse:maxiter.

En cada paso iterativo, el valor esperado del hamiltoniano se estima mediante muchas mediciones. Este valor de energía estimado es devuelto por la función de costo, y el minimizador actualiza su información sobre el paisaje energético. La forma exacta en que el optimizador elige el siguiente paso depende del método. Algunos utilizan gradientes y eligen la dirección de descenso más pronunciado. Otros tienen en cuenta el ruido y requieren que el costo disminuya por un margen considerable antes de aceptar que la energía verdadera disminuye en esa dirección.
# Example syntax for minimization
# from scipy.optimize import minimize
# res = minimize(cost_func, x0, args=(ansatz, hamiltonian, estimator), method="cobyla",
# options={'maxiter': 200})
1.5 El principio variacional
El principio variacional es de importancia central en este contexto: establece que ninguna función de onda variacional puede producir un valor esperado de energía (o costo) menor que el de la función de onda del estado fundamental. Expresado matemáticamente:
Esto se puede demostrar fácilmente si se observa que el conjunto de todos los estados propios de forma una base completa para el espacio de Hilbert. En otras palabras: cualquier estado, en particular , puede escribirse como una suma ponderada (normalizada) de estos estados propios de :
donde son constantes por determinar y se cumple que . Dejamos esto como ejercicio. Sin embargo, observe la consecuencia: el estado variacional que produce el menor valor esperado de la energía es la mejor aproximación del verdadero estado fundamental.
Verifica tu comprensión
Demuestre matemáticamente que para cualquier estado variacional .
Respuesta
Con la expansión dada del estado variacional en estados propios de energía,
podemos escribir el valor esperado de la energía variacional como
Para todos los coeficientes se cumple . De esto se obtiene:
2. Comparación con el flujo de trabajo clásico
Suponga que le interesa una matriz con N filas y N columnas. Imagine que su matriz es tan grande que la diagonalización exacta no es una opción. Suponga que conoce lo suficiente sobre su problema como para hacer algunas suposiciones sobre la estructura general del estado propio objetivo, y desea explorar estados similares a su estimación inicial para ver si su costo/energía puede reducirse aún más. Este es un enfoque variacional y un método que se emplea cuando la diagonalización exacta no es una opción.
2.1 Flujo de trabajo clásico
Con un computador clásico, esto funcionaría de la siguiente manera:
- Construya un estado estimado con algunos parámetros que variará: . Aunque esta estimación inicial podría ser aleatoria, no es recomendable. Queremos aprovechar el conocimiento sobre el problema en cuestión para ajustar nuestra estimación tanto como sea posible.
- Calcule el valor esperado del operador cuando el sistema se encuentra en este estado:
- Cambie los parámetros variacionales y repita: .
- Utilice la información recopilada sobre el paisaje de estados posibles en su subespacio variacional para hacer estimaciones cada vez mejores y acercarse al estado objetivo. El principio variacional garantiza que nuestro estado variacional no puede producir un valor propio menor que el del estado fundamental buscado. Por lo tanto, cuanto menor sea el valor esperado, mejor será nuestra aproximación del estado fundamental:
Consideremos la dificultad de cada paso en este enfoque. Establecer o actualizar parámetros es computacionalmente sencillo; la dificultad radica en elegir parámetros iniciales útiles y físicamente motivados. Utilizar la información recopilada de iteraciones anteriores para actualizar los parámetros de modo que nos acerquemos al estado fundamental no es trivial. Sin embargo, existen algoritmos de optimización clásica que lo hacen de manera bastante eficiente. Esta optimización clásica solo es costosa porque puede requerir muchas iteraciones; en el peor caso, el número de iteraciones puede escalar exponencialmente con N. El paso individual computacionalmente más intenso es, con gran probabilidad, el cálculo del valor esperado de su matriz con un estado dado :
La matriz de debe actuar sobre el vector de elementos, lo que en el peor caso corresponde a operaciones de multiplicación. Esto debe realizarse en cada iteración de parámetros. Para matrices extremadamente grandes, esto conlleva un alto costo computacional.
2.2 Flujo de trabajo cuántico y grupos de Pauli conmutantes
Imagine ahora que transfiere esta parte del cálculo a un computador cuántico. En lugar de calcular este valor esperado, lo estima preparando el estado en el computador cuántico con su ansatz variacional y luego realizando mediciones.
Esto suena más sencillo de lo que es. en general no es fácil de medir. Podría, por ejemplo, estar compuesto de muchos operadores de Pauli X, Y y Z que no conmutan. Pero puede escribirse como una combinación lineal de términos , cada uno de los cuales es fácilmente medible (por ejemplo, operadores de Pauli o grupos de operadores de Pauli que conmutan qubit a qubit). El valor esperado de sobre un estado es la suma ponderada de los valores esperados de los términos constituyentes . Esta expresión es válida para cualquier estado , pero la utilizaremos específicamente con nuestros estados variacionales .
donde es una cadena de Pauli como IZZX…XIYX, o varias de estas cadenas que conmutan entre sí. Una descripción del valor esperado que se acerca más a la realidad de las mediciones en computadores cuánticos es, por lo tanto:
Y en el contexto de nuestra función de onda variacional:
Cada uno de los términos puede medirse veces y produce muestras de medición con , así como un valor esperado y una desviación estándar . Estos términos pueden sumarse y los errores propagarse a través de la suma para obtener un valor esperado total y una desviación estándar total .
Esto no requiere multiplicaciones de matrices costosas ni un proceso que necesariamente escale como . En cambio, se requieren múltiples mediciones en el computador cuántico. Si no se necesitan demasiadas, este enfoque podría ser eficiente. Esta es la parte cuántica de VQE.
Sin embargo, hablemos de las razones por las que esto podría no ser eficiente. Una razón para muchas mediciones es la reducción de la incertidumbre estadística en las estimaciones, necesaria para cálculos muy precisos. Otra razón es la cantidad de cadenas de Pauli necesarias para cubrir toda la matriz. Dado que las matrices de Pauli (más la identidad: X, Y, Z e I) generan el espacio de todos los operadores de una dimensión dada, está garantizado que podemos escribir nuestra matriz de interés como una suma ponderada de operadores de Pauli, como hicimos anteriormente.
donde es una cadena de Pauli que actúa sobre todos los qubits del sistema, como IZZX…XIYX, o varias de estas cadenas que conmutan entre sí. Nota que Qiskit utiliza la notación little-endian, donde el operador de Pauli desde la derecha actúa sobre el qubit. Por lo tanto, podemos medir nuestro operador midiendo una serie de operadores de Pauli.
Sin embargo, no podemos medir todos estos operadores de Pauli simultáneamente. Los operadores de Pauli (sin I) no conmutan entre sí cuando están asociados al mismo qubit. Por ejemplo, podemos medir IZIZ y ZZXZ simultáneamente, porque podemos medir I y Z al mismo tiempo para el tercer qubit, e I y X al mismo tiempo para el primer qubit. Pero no podemos medir ZZZZ y ZZZX simultáneamente, porque Z y X no conmutan y ambos actúan sobre el qubit 0. Los lectores con experiencia pueden recordar que dos grupos de operadores de Pauli pueden conmutar como conjunto, aunque las mediciones de cada qubit individual no conmuten. Estimator asume mediciones de Pauli en producto tensorial (mediante rotaciones de base), lo que corresponde a agrupar operadores que conmutan qubit a qubit. Por tanto, para estimar simultáneamente dos cadenas (A y B) de operadores de Pauli usando Estimator, los operadores de Pauli de cada qubit en A y B deben conmutar. Esto significa que tampoco podemos medir ZZZZ y ZZXX simultáneamente.

Por lo tanto, descomponemos nuestra matriz en una suma de operadores de Pauli que actúan sobre diferentes qubits. Algunos elementos de esta suma pueden medirse simultáneamente; a esto lo llamamos un grupo de Paulis conmutantes. Dependiendo de cuántos términos no conmutantes haya, podríamos necesitar muchos de estos grupos. Al número de tales grupos de cadenas de Pauli conmutantes lo llamamos . Si es pequeño, esto podría funcionar bien. Si tiene millones de grupos, esto no será útil.
Los procesos necesarios para la estimación del valor esperado están encapsulados en la primitiva Estimator del entorno de ejecución de Qiskit. Puede obtener más información sobre el Estimator en la referencia de API en la documentación de IBM Quantum®. Aunque se puede utilizar el Estimator directamente, este devuelve más que solo el valor propio de energía más pequeño, por ejemplo, también información sobre el error estándar del ensemble. En el contexto de problemas de minimización, el Estimator se utiliza frecuentemente dentro de una función de costo. Puede obtener más información sobre las entradas y salidas del Estimator en esta guía en la documentación de IBM Quantum.
Se registra el valor esperado (o la función de costo) para el conjunto de parámetros utilizado en su estado, y luego se actualizan los parámetros. Con el tiempo, podría utilizar los valores esperados o los valores de la función de costo estimados para aproximar un gradiente de su función de costo en el subespacio de estados muestreados por su ansatz. Existen tanto optimizadores clásicos basados en gradientes como libres de gradientes. Ambos pueden sufrir problemas de entrenabilidad, como múltiples mínimos locales y grandes regiones del espacio de parámetros con gradiente cercano a cero, conocidas como barren plateaus.

2.3 Factores que determinan el costo computacional
VQE no resolverá todos sus problemas más difíciles de química cuántica. Eso es cierto. Pero ser mejor en todos los cálculos tampoco es el objetivo. Hemos cambiado lo que determina el costo computacional.

Hemos pasado de un proceso cuya complejidad depende solo de la dimensión de la matriz a un proceso que depende de la precisión requerida y del número de operadores de Pauli no conmutantes que componen la matriz. Este último aspecto no tiene análogo en la computación clásica.
Basándose en estas dependencias, este proceso podría ser útil para matrices dispersas o matrices con pocas cadenas de Pauli no conmutantes. Este es el caso, por ejemplo, de sistemas de espines interactuantes. Para matrices densas, podría ser menos útil. Sabemos, por ejemplo, que los sistemas químicos a menudo tienen hamiltonianos que comprenden cientos, miles o incluso millones de cadenas de Pauli. Existen trabajos interesantes para reducir este número de términos. Pero los sistemas químicos podrían adaptarse mejor a algunos de los otros algoritmos que discutiremos en este curso.
Verifica tu comprensión
Considere un hamiltoniano de cuatro qubits que contiene los siguientes términos:
IIXX, IIXZ, IIZZ, IZXZ, IXXZ, ZZXZ, XZXZ, ZIXZ, ZZZZ, XXXX
Desea ordenar estos términos en grupos de modo que todos los términos de un grupo puedan medirse simultáneamente. ¿Cuál es el número mínimo de tales grupos que puede formar para que todos los términos estén cubiertos?
Respuesta
Es posible hacerlo en 4 grupos. Nota que estas soluciones típicamente no son únicas.
IIXX, XXXX, IIZZ, ZZZZ
IIXZ, IZXZ, ZIXZ, ZZXZ
IXXZ
XZXZ
¿Qué espera que haga típicamente difícil la química cuántica con VQE: la cantidad de términos en el hamiltoniano o encontrar un buen ansatz?
Respuesta
Existen ansätze que están altamente optimizados para contextos químicos. La cantidad de términos en el hamiltoniano y, por lo tanto, la cantidad de mediciones requeridas, típicamente causan más problemas.
3. Hamiltoniano de ejemplo
Pongamos en práctica este algoritmo con un hamiltoniano pequeño para poder ver qué sucede en cada paso. Utilizamos el marco de trabajo Qiskit Patterns:
-Paso 1: Mapear el problema a circuitos cuánticos y operadores -Paso 2: Optimizar para el hardware objetivo -Paso 3: Ejecutar en el hardware objetivo -Paso 4: Posprocesar resultados
3.1 Paso 1: Mapear el problema a circuitos cuánticos y operadores
Utilizamos el hamiltoniano definido anteriormente en el contexto químico. Primero, algunas importaciones generales.
# General imports
import numpy as np
# SciPy minimizer routine
from scipy.optimize import minimize
# Plotting functions
import matplotlib.pyplot as plt
Asumimos que el hamiltoniano buscado es conocido. Aquí utilizamos un hamiltoniano muy pequeño, ya que otros métodos discutidos en este curso pueden resolver problemas más grandes de manera más eficiente.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
hamiltonian = SparsePauliOp.from_list(
[("YZ", 0.3980), ("ZI", -0.3980), ("ZZ", -0.0113), ("XX", 0.1810)]
)
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues))
The ground state energy is -0.702930394459531
En Qiskit hay muchas opciones de ansatz predefinidas. Utilizamos efficient_su2.
# Pre-defined ansatz circuit and operator class for Hamiltonian
from qiskit.circuit.library import efficient_su2
# Note that it is more common to place initial 'h' gates outside the ansatz.
# Here we specifically wanted this layer structure.
ansatz = efficient_su2(
hamiltonian.num_qubits, su2_gates=["h", "rz", "y"], entanglement="circular", reps=1
)
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.decompose().draw("mpl", style="iqp")
This circuit has 4 parameters
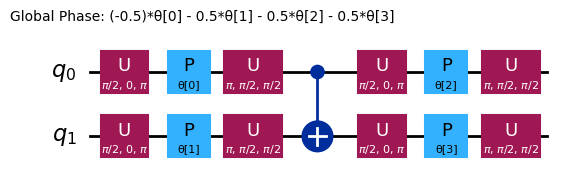
Diferentes ansätze tienen diferentes estructuras de entrelazamiento y diferentes puertas de rotación. El que se muestra aquí utiliza puertas CNOT para el entrelazamiento, así como puertas Y y puertas RZ parametrizadas para las rotaciones. Nota el tamaño de este espacio de parámetros: debemos minimizar la función de costo sobre 4 variables (los parámetros de las puertas RZ). Esto puede escalar, pero no indefinidamente. Un problema similar en 4 qubits con las 3 repeticiones estándar de efficient_su2 produce 16 parámetros variacionales.
3.2 Paso 2: Optimizar para el hardware objetivo
El ansatz se escribió con puertas conocidas, pero nuestro circuito debe transpilarse para poder utilizar las puertas base implementadas en el computador cuántico específico. Seleccionamos el backend menos ocupado.
# runtime imports
from qiskit_ibm_runtime import QiskitRuntimeService, Session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# To run on hardware, select the backend with the fewest number of jobs in the queue
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
print(backend)
<IBMBackend('ibm_torino')>
Ahora podemos transpilar nuestro circuito para este hardware y visualizar el ansatz transpilado.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")

Nota que las puertas utilizadas han cambiado y los qubits de nuestro circuito abstracto se han mapeado a qubits con diferente numeración en el computador cuántico. Para obtener resultados significativos, debemos mapear nuestro hamiltoniano de la misma manera.
hamiltonian_isa = hamiltonian.apply_layout(layout=ansatz_isa.layout)
3.3 Paso 3: Ejecutar en el hardware objetivo
3.3.1 Reportar valores
Aquí definimos una función de costo que acepta como argumentos las estructuras creadas en los pasos anteriores: los parámetros, el ansatz y el hamiltoniano. También utiliza el Estimator, que aún no hemos definido. Para verificar el comportamiento de convergencia, añadimos código que registra el historial de la función de costo.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from Estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
Es muy ventajoso si puede elegir los valores iniciales de los parámetros basándose en el conocimiento del problema particular y las propiedades del estado objetivo. No hacemos tales suposiciones y utilizamos valores iniciales aleatorios.
x0 = 2 * np.pi * np.random.random(num_params)
# This required 13 min, 20 s QPU time on an Eagle processor, 28 min total time.
with Session(backend=backend) as session:
estimator = Estimator(mode=session)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 50},
)
Iters. done: 1 [Current cost: 0.010575798722044727]
Iters. done: 2 [Current cost: 0.004040015974440895]
Iters. done: 3 [Current cost: 0.0020213258785942503]
Iters. done: 4 [Current cost: 0.18723082446726014]
Iters. done: 5 [Current cost: -0.2746792152068885]
Iters. done: 6 [Current cost: -0.3094547651648519]
Iters. done: 7 [Current cost: -0.05281985428356641]
Iters. done: 8 [Current cost: 0.00808560303514377]
Iters. done: 9 [Current cost: -0.0014821685303514388]
Iters. done: 10 [Current cost: -0.004759824281150161]
Iters. done: 11 [Current cost: 0.09942328705995292]
Iters. done: 12 [Current cost: 0.01092366214057508]
Iters. done: 13 [Current cost: 0.05017497496069776]
Iters. done: 14 [Current cost: 0.13028868414310696]
Iters. done: 15 [Current cost: 0.013747803514376994]
Iters. done: 16 [Current cost: 0.2583072432944498]
Iters. done: 17 [Current cost: -0.14422125655131562]
Iters. done: 18 [Current cost: -0.0004950150347678081]
Iters. done: 19 [Current cost: 0.00681082268370607]
Iters. done: 20 [Current cost: -0.0023377795527156544]
Iters. done: 21 [Current cost: 0.6027665591169237]
Iters. done: 22 [Current cost: 0.00596641373801917]
Iters. done: 23 [Current cost: -0.008318769968051117]
Iters. done: 24 [Current cost: -0.00026683306709265246]
Iters. done: 25 [Current cost: -0.007648222843450479]
Iters. done: 26 [Current cost: 0.004121086261980831]
Iters. done: 27 [Current cost: -0.004075019968051117]
Iters. done: 28 [Current cost: -0.004419369009584665]
Iters. done: 29 [Current cost: 0.213185460054037]
Iters. done: 30 [Current cost: -0.06505919572162797]
Iters. done: 31 [Current cost: -0.5334241316590271]
Iters. done: 32 [Current cost: 0.00218370607028754]
Iters. done: 33 [Current cost: 0.09579352143666908]
Iters. done: 34 [Current cost: -0.009274800319488819]
Iters. done: 35 [Current cost: -0.44395141360688106]
Iters. done: 36 [Current cost: 0.011747104632587858]
Iters. done: 37 [Current cost: -0.003344149361022364]
Iters. done: 38 [Current cost: 0.19138183916486304]
Iters. done: 39 [Current cost: 0.013513931813145209]
Podemos examinar las salidas sin procesar.
res
message: Return from COBYLA because the trust region radius reaches its lower bound.
success: True
status: 0
fun: -0.5334241316590271
x: [ 1.024e+00 6.459e+00 3.625e+00 4.007e+00]
nfev: 39
maxcv: 0.0
3.4 Paso 4: Posprocesar resultados
Si el procedimiento termina correctamente, los valores en nuestro diccionario deberían corresponder al vector solución y al número total de evaluaciones de la función, respectivamente. Esto se puede verificar fácilmente:
cost_history_dict
{'prev_vector': array([1.02397956, 6.45886604, 3.62479262, 4.00744128]),
'iters': 39,
'cost_history': [np.float64(0.010575798722044727),
np.float64(0.004040015974440895),
np.float64(0.0020213258785942503),
np.float64(0.18723082446726014),
np.float64(-0.2746792152068885),
np.float64(-0.3094547651648519),
np.float64(-0.05281985428356641),
np.float64(0.00808560303514377),
np.float64(-0.0014821685303514388),
np.float64(-0.004759824281150161),
np.float64(0.09942328705995292),
np.float64(0.01092366214057508),
np.float64(0.05017497496069776),
np.float64(0.13028868414310696),
np.float64(0.013747803514376994),
np.float64(0.2583072432944498),
np.float64(-0.14422125655131562),
np.float64(-0.0004950150347678081),
np.float64(0.00681082268370607),
np.float64(-0.0023377795527156544),
np.float64(0.6027665591169237),
np.float64(0.00596641373801917),
np.float64(-0.008318769968051117),
np.float64(-0.00026683306709265246),
np.float64(-0.007648222843450479),
np.float64(0.004121086261980831),
np.float64(-0.004075019968051117),
np.float64(-0.004419369009584665),
np.float64(0.213185460054037),
np.float64(-0.06505919572162797),
np.float64(-0.5334241316590271),
np.float64(0.00218370607028754),
np.float64(0.09579352143666908),
np.float64(-0.009274800319488819),
np.float64(-0.44395141360688106),
np.float64(0.011747104632587858),
np.float64(-0.003344149361022364),
np.float64(0.19138183916486304),
np.float64(0.013513931813145209)]}
fig, ax = plt.subplots()
x = np.linspace(0, 10, 50)
# Define the constant function
constant = -0.7029
y_constant = np.full_like(x, constant)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

IBM Quantum ofrece recursos de aprendizaje adicionales sobre VQE. Si desea poner VQE en práctica, consulte nuestro tutorial: Estimación de la energía del estado fundamental de la cadena de Heisenberg con VQE. Para más información sobre la creación de hamiltonianos moleculares, consulte esta lección de nuestro curso sobre Química cuántica con VQE. Para una comprensión más profunda del funcionamiento de algoritmos variacionales como VQE, recomendamos el curso Variational Algorithm Design.
Verifica tu comprensión
En esta sección hemos calculado una energía del estado fundamental a partir de un hamiltoniano. ¿Cómo procederíamos si quisiéramos aplicar esto a la determinación de la geometría de una molécula?
Respuesta
Tendríamos que introducir variables para las distancias interatómicas y los ángulos entre los enlaces, y variarlas. Para cada variación de estas magnitudes surgiría un nuevo hamiltoniano (ya que los operadores que describen la energía dependen de la geometría). Para cada hamiltoniano así generado y mapeado a qubits, habría que realizar una optimización como la descrita anteriormente. De todos estos problemas de optimización convergidos, la geometría con la energía más baja sería la que se encuentra en la naturaleza. Esto es considerablemente más costoso que lo mostrado arriba. Tal cálculo se realiza para la molécula más simple, , aquí.
4. Relación de VQE con otros métodos
En esta sección consideramos las ventajas y desventajas del enfoque original de VQE y mostramos sus relaciones con otros algoritmos más recientes.
4.1 Las fortalezas y debilidades de VQE
Algunas fortalezas ya se han mencionado. Estas incluyen:
- Idoneidad para el hardware actual: Algunos algoritmos cuánticos requieren tasas de error significativamente más bajas, cercanas a la tolerancia a fallos a gran escala. VQE no; puede implementarse en los computadores cuánticos actuales.
- Circuitos poco profundos: VQE utiliza frecuentemente circuitos cuánticos relativamente poco profundos. Esto hace que VQE sea menos susceptible a errores de puertas acumulados y adecuado para muchas técnicas de mitigación de errores. Por supuesto, los circuitos no siempre son poco profundos; esto depende del ansatz utilizado.
- Versatilidad: VQE puede (en principio) aplicarse a cualquier problema que pueda formularse como un problema de valores propios/vectores propios. Sin embargo, existen muchas limitaciones que hacen que VQE sea poco práctico o desventajoso para algunos problemas. Algunas de ellas se resumen a continuación.
Algunas debilidades de VQE y problemas para los que resulta poco práctico ya se han descrito anteriormente. Estas incluyen:
- Naturaleza heurística: VQE no garantiza la convergencia a la energía correcta del estado fundamental, ya que su rendimiento depende de la elección del ansatz y los métodos de optimización[1-2]. Si se elige un ansatz inadecuado que carece del entrelazamiento necesario para el estado fundamental deseado, ningún optimizador clásico podrá alcanzar dicho estado fundamental.
- Potencialmente muchos parámetros: Un ansatz muy expresivo puede tener tantos parámetros que las iteraciones de minimización se vuelvan muy lentas.
- Alto costo de medición: En VQE se utiliza un Estimator para estimar el valor esperado de cada término en el hamiltoniano. Para la mayoría de los hamiltonianos relevantes, hay términos que no pueden estimarse simultáneamente. Esto puede hacer que VQE sea intensivo en recursos para sistemas grandes con hamiltonianos complicados[1].
- Efectos del ruido: Cuando el optimizador clásico busca un mínimo, los cálculos ruidosos pueden confundirlo y alejarlo del verdadero mínimo o retrasar la convergencia. Una posible solución para esto es el empleo de técnicas de vanguardia de mitigación y supresión de errores[2-3] de IBM.
- Barren plateaus: Estas regiones de gradientes que se desvanecen[2-3] existen incluso sin ruido, pero se vuelven más problemáticas con ruido, ya que los cambios en los valores esperados causados por el ruido pueden ser mayores que los cambios debidos a la actualización de parámetros en estas regiones planas.
4.2 Relación con otros enfoques
Adapt-VQE
El algoritmo ADAPT-VQE (Adaptive Derivative-Assembled Pseudo-Trotter Variational Quantum Eigensolver) es una evolución del algoritmo VQE original que busca mejorar la eficiencia, precisión y escalabilidad en simulaciones cuánticas, especialmente en química cuántica.
El algoritmo VQE original, descrito en esta lección, utiliza un ansatz predefinido y fijo para aproximarse al estado fundamental del sistema. En nuestro caso, utilizamos efficient_su2 con una sola repetición y puertas de rotación Y y RZ. Aunque los parámetros de las puertas RZ cambiaron, la estructura y las puertas de este ansatz permanecieron inalteradas.
ADAPT-VQE aborda las limitaciones de VQE mediante la construcción adaptativa del ansatz. En lugar de comenzar con un ansatz fijo, ADAPT-VQE construye el ansatz de forma dinámica e iterativa. En cada paso, se selecciona del conjunto predefinido (por ejemplo, operadores de excitación fermiónicos) el operador que presenta el mayor gradiente respecto a la energía. De esta manera, solo se añaden los operadores más impactantes, lo que resulta en un ansatz compacto y eficiente[4-6]. Este enfoque puede tener varios efectos beneficiosos:
- Profundidad de circuito reducida: Mediante el crecimiento incremental del ansatz y el enfoque en los operadores necesarios, ADAPT-VQE minimiza las operaciones de puertas en comparación con los enfoques VQE convencionales[5,7].
- Precisión mejorada: La naturaleza adaptativa permite a ADAPT-VQE capturar más energía de correlación en cada paso, lo que lo hace especialmente efectivo para sistemas fuertemente correlacionados donde el VQE tradicional tiene dificultades[8,9].
- Escalabilidad y robustez frente al ruido: El ansatz compacto reduce la acumulación de puertas, el costo computacional y la cantidad de parámetros variacionales a minimizar.
Sin embargo, ADAPT-VQE no es perfecto. En algunos casos puede quedar atrapado en mínimos locales o ralentizarse, y puede sufrir de sobreparametrización. Además, puede ser bastante intensivo en recursos, ya que requiere el cálculo de gradientes y la optimización de parámetros con muchas estructuras de puertas.
Estimación de fase cuántica (QPE)
QPE persigue un propósito similar al de VQE, pero difiere considerablemente en su implementación. QPE requiere computadores cuánticos tolerantes a fallos debido a sus circuitos cuánticos generalmente profundos y al alto grado de coherencia que necesita. Una vez que QPE pueda implementarse, sería más preciso que VQE. Una forma de describir la diferencia es la precisión como función de la profundidad del circuito. QPE alcanza la precisión con una profundidad de circuito que escala como [10]. VQE necesita mediciones para alcanzar la misma precisión[10,11].
Krylov, SQD, QSCI y otros en este curso
VQE ha contribuido a establecer algoritmos cuánticos que siguen dependiendo de computadores clásicos, no solo para operar el computador cuántico, sino para partes esenciales del algoritmo. Varios de estos algoritmos son el foco del resto del curso. Aquí ofrecemos una breve descripción general de algunos de ellos para compararlos con VQE. En las lecciones siguientes se explicarán con mucho más detalle.
Diagonalización cuántica de Krylov (KQD)
Los métodos de subespacio de Krylov son procedimientos para proyectar una matriz sobre un subespacio, reducir su dimensión y hacerla manejable mientras se conservan las propiedades más importantes. Un aspecto clave de este método consiste en generar un subespacio que preserve estas propiedades; la generación de este subespacio está estrechamente relacionada con un método establecido en computadores cuánticos, la llamada trotterización.
Existen algunas variantes de los métodos cuánticos de Krylov, pero en general el enfoque es:
- Utilizar el computador cuántico para generar un subespacio (el subespacio de Krylov) mediante trotterización
- Proyectar la matriz de interés sobre este subespacio de Krylov
- Diagonalizar el nuevo hamiltoniano proyectado con un computador clásico
Diagonalización cuántica basada en muestreo (SQD)
La diagonalización cuántica basada en muestreo (SQD) está relacionada con el método de Krylov, ya que también intenta reducir la dimensión de una matriz a diagonalizar mientras conserva propiedades importantes. SQD procede de la siguiente manera:
- Comenzar con una estimación inicial del estado fundamental y preparar el sistema en dicho estado fundamental.
- Utilizar el Sampler para muestrear las cadenas de bits que componen este estado.
- Usar la colección de estados de la base computacional del Sampler como subespacio sobre el cual se proyecta la matriz de interés.
- Diagonalizar la matriz proyectada, más pequeña, con un computador clásico.
Esto está relacionado con VQE en que utiliza computación clásica y cuántica para componentes esenciales del algoritmo. Ambos también comparten el requisito de tener una buena estimación inicial o un buen ansatz. Pero la distribución del trabajo entre el computador clásico y el cuántico en SQD se asemeja más a la del método de Krylov.
De hecho, el método de Krylov y SQD se han combinado recientemente en el método de diagonalización cuántica de Krylov basada en muestreo (SKQD)[12].
Interacción de configuraciones seleccionada cuánticamente
La Quantum Selected Configuration Interaction (QSCI)[13] es un algoritmo que genera un estado fundamental aproximado de un hamiltoniano muestreando una función de onda de prueba para identificar los estados de la base computacional más significativos y así generar un subespacio para una diagonalización clásica. Tanto SQD como QSCI utilizan un computador cuántico para construir un subespacio reducido. La fortaleza particular de QSCI reside en su preparación de estados, especialmente en el contexto de problemas químicos. Utiliza diversas estrategias como estados evolucionados en el tiempo[14] y una serie de ansätze inspirados en la química. Al enfocarse en la preparación eficiente de estados, QSCI reduce los costos de computación cuántica para hamiltonianos químicos, mientras mantiene una alta precisión y aprovecha la robustez frente al ruido mediante técnicas de muestreo de estados cuánticos[15]. QSCI también ofrece una técnica de construcción adaptativa que proporciona más ansätze para mejores resultados.
El flujo de trabajo estándar de QSCI para problemas químicos es el siguiente:
- Construir el hamiltoniano molecular con un software de su elección (por ejemplo, SciPy).
- Preparar un algoritmo QSCI eligiendo un estado inicial apropiado y un ansatz inspirado en la química con un conjunto de parámetros preseleccionado.
- Muestrear estados base significativos y diagonalizar el hamiltoniano con un computador clásico para obtener la energía del estado fundamental.
- Frecuentemente se emplean la reconstrucción de configuraciones[16] y la postselección por simetría[15] como técnicas de posprocesamiento.
- Opcionalmente, el flujo de trabajo del QSCI adaptativo tiene un bucle de optimización adicional del paso 2 al paso 3, utilizando más ansätze con estados iniciales aleatorios.
Verifica tu comprensión
¿Qué tiene VQE en común con todos los demás métodos enumerados anteriormente (excepto QPE, que no se describe en detalle)?
Respuesta
Todos utilizan un estado de prueba o una función de onda de algún tipo. Todos funcionan mejor cuando la estimación inicial para este estado de prueba es excelente.
Otra respuesta correcta es que todos son más fáciles de implementar cuando el hamiltoniano es fácil de medir (es decir, puede clasificarse en relativamente pocos grupos de operadores de Pauli conmutantes).
¿Qué tiene VQE en común con ninguno de los otros métodos enumerados anteriormente?
Respuesta
Optimizadores clásicos. Ninguno de los otros utiliza algoritmos de optimización clásica para seleccionar los parámetros variacionales.
Referencias
[2] https://en.wikipedia.org/wiki/Variational_quantum_eigensolver
[3] https://journals.aps.org/prapplied/abstract/10.1103/PhysRevApplied.19.024047
[4] https://arxiv.org/abs/2111.05176
[6] https://inquanto.quantinuum.com/tutorials/InQ_tut_fe4n2_2.html
[7] https://www.nature.com/articles/s41467-019-10988-2
[8] https://arxiv.org/abs/2210.15438
[9] https://journals.aps.org/prresearch/abstract/10.1103/PhysRevResearch.6.013254
[10] https://arxiv.org/html/2403.09624v1
[11] https://www.nature.com/articles/s42005-023-01312-y
[13] https://arxiv.org/abs/1802.00171
[14] https://arxiv.org/abs/2103.08505
[15] https://arxiv.org/html/2501.09702v1
[16] https://quri-sdk.qunasys.com/docs/examples/quri-algo-vm/qsci/
[17] https://arxiv.org/abs/2412.13839