Diagonalización cuántica de Krylov
En esta lección sobre la diagonalización cuántica de Krylov (KQD) responderemos las siguientes preguntas:
- ¿Qué es el método de Krylov, en general?
- ¿Por qué funciona el método de Krylov y bajo qué condiciones?
- ¿Qué papel juega la computación cuántica?
La parte cuántica de los cálculos se basa principalmente en el trabajo de la Ref [1].
El video a continuación ofrece una visión general de los métodos de Krylov en la computación clásica, motiva su uso y explica cómo la computación cuántica puede desempeñar un papel en ese flujo de trabajo. El texto posterior ofrece más detalles e implementa un método de Krylov tanto de forma clásica como usando un computador cuántico.
1. Introducción a los métodos de Krylov
Un método de subespacios de Krylov puede referirse a cualquiera de varios métodos construidos alrededor de lo que se denomina el subespacio de Krylov. Una revisión completa de estos métodos queda fuera del alcance de esta lección, pero las Refs [2-4] ofrecen una base mucho más completa. Aquí nos centraremos en qué es un subespacio de Krylov, cómo y por qué resulta útil para resolver problemas de valores propios, y finalmente cómo puede implementarse en un computador cuántico.
Definición: Dada una matriz simétrica y semidefinida positiva , el espacio de Krylov de orden es el espacio generado por los vectores obtenidos al multiplicar potencias crecientes de la matriz , hasta , con un vector de referencia .
Aunque los vectores anteriores generan lo que llamamos un subespacio de Krylov, no hay razón para pensar que serán ortogonales. Frecuentemente se utiliza un proceso iterativo de ortonormalización similar a la ortogonalización de Gram-Schmidt. Aquí el proceso es ligeramente diferente: cada nuevo vector se hace ortogonal a los demás a medida que se genera. En este contexto, esto se denomina iteración de Arnoldi. Partiendo del vector inicial , se genera el siguiente vector y luego se asegura que este segundo vector sea ortogonal al primero restando su proyección sobre . Es decir:
Ahora es fácil ver que ya que
Hacemos lo mismo para el siguiente vector, asegurándonos de que sea ortogonal a los dos anteriores:
Si repetimos este proceso para los vectores, tenemos una base ortonormal completa para un espacio de Krylov. Nótese que el proceso de ortogonalización dará cero en cuanto , ya que vectores ortogonales necesariamente abarcan el espacio completo. El proceso también dará cero si algún vector es un vector propio de , ya que todos los vectores siguientes serán múltiplos de ese vector.
1.1 Un ejemplo sencillo: Krylov a mano
Repasemos paso a paso la generación de un subespacio de Krylov sobre una matriz trivialmente pequeña, para poder ver el proceso en detalle. Comenzamos con una matriz inicial de nuestro interés:
Para este pequeño ejemplo, podemos determinar los vectores propios y los valores propios fácilmente incluso a mano. Aquí mostramos la solución numérica.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy sympy
# One might use linalg.eigh here, but later matrices may not be Hermitian. So we use
# linalg.eig in this lesson.
import numpy as np
A = np.array([[4, -1, 0], [-1, 4, -1], [0, -1, 4]])
eigenvalues, eigenvectors = np.linalg.eig(A)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
The eigenvalues are [2.58578644 4. 5.41421356]
The eigenvectors are [[ 5.00000000e-01 -7.07106781e-01 5.00000000e-01]
[ 7.07106781e-01 1.37464400e-16 -7.07106781e-01]
[ 5.00000000e-01 7.07106781e-01 5.00000000e-01]]
Los registramos aquí para comparación posterior:
Nos gustaría estudiar cómo funciona este proceso (o falla) a medida que aumentamos la dimensión de nuestro subespacio de Krylov, . Con este fin, aplicaremos el siguiente proceso:
- Generar un subespacio del espacio vectorial completo comenzando con un vector elegido aleatoriamente (llamarlo si ya está normalizado, como arriba).
- Proyectar la matriz completa sobre ese subespacio y encontrar los valores propios de esa matriz proyectada .
- Aumentar el tamaño del subespacio generando más vectores, asegurando que sean ortonormales, usando un proceso similar a la ortogonalización de Gram-Schmidt.
- Proyectar sobre el subespacio más grande y encontrar los valores propios de la matriz resultante .
- Repetir hasta que los valores propios converjan (o en este caso de juguete, hasta haber generado vectores que abarquen el espacio vectorial completo de la matriz original ).
Una implementación normal del método de Krylov no necesitaría resolver el problema de valores propios para la matriz proyectada sobre cada subespacio de Krylov a medida que se construye. Podrías construir el subespacio de la dimensión deseada, proyectar la matriz sobre ese subespacio y diagonalizar la matriz proyectada. Proyectar y diagonalizar en cada dimensión del subespacio solo se hace para verificar la convergencia.
Dimensión :
Elegimos un vector aleatorio, digamos
Si aún no está normalizado, normalízalo.
Ahora proyectamos nuestra matriz sobre el subespacio de este único vector:
Esta es nuestra proyección de la matriz sobre el subespacio de Krylov cuando solo contiene un único vector, . El valor propio de esta matriz es trivialmente 4. Podemos pensar en esto como nuestra estimación de orden cero de los valores propios (en este caso solo uno) de . Aunque es una estimación pobre, es del orden de magnitud correcto.
Dimensión :
Ahora generamos el siguiente vector en nuestro subespacio mediante la operación de sobre el vector anterior:
Ahora restamos la proyección de este vector sobre nuestro vector anterior para garantizar la ortogonalidad.
Si aún no está normalizado, normalízalo. En este caso, el vector ya estaba normalizado, por lo que
Ahora proyectamos nuestra matriz A sobre el subespacio de estos dos vectores:
Aún nos queda el problema de determinar los valores propios de esta matriz. Pero esta matriz es ligeramente más pequeña que la matriz completa. En problemas con matrices muy grandes, trabajar con este subespacio más pequeño puede ser muy ventajoso.
Aunque esta sigue siendo una estimación pobre, es mejor que la de orden cero. Llevaremos esto a cabo una iteración más para asegurarnos de que el proceso queda claro. Sin embargo, esto socava el propósito del método, ya que terminaremos diagonalizando una matriz 3x3 en la siguiente iteración, lo que significa que no hemos ahorrado tiempo ni potencia computacional.
Dimensión :
Ahora generamos el siguiente vector en nuestro subespacio mediante la operación de A sobre el vector anterior:
Ahora restamos la proyección de este vector sobre ambos vectores anteriores para garantizar la ortogonalidad.
Si aún no está normalizado, normalízalo. En este caso, el vector ya estaba normalizado, por lo que
Ahora proyectamos nuestra matriz sobre el subespacio de estos vectores:
Ahora determinamos los valores propios:
Estos valores propios son exactamente los valores propios de la matriz original . Esto debe ser así, ya que hemos expandido nuestro subespacio de Krylov para abarcar el espacio vectorial completo de la matriz original .
En este ejemplo, el método de Krylov puede no parecer particularmente más sencillo que la diagonalización directa. De hecho, como veremos en secciones posteriores, el método de Krylov solo es ventajoso por encima de cierta dimensión de la matriz; está pensado para ayudarnos a resolver problemas de valores/vectores propios de matrices extremadamente grandes.
Este es el único ejemplo que mostraremos resuelto "a mano", pero la sección 2 a continuación muestra ejemplos computacionales.
Aclaración de términos
Un malentendido común es que solo existe un único subespacio de Krylov para un problema dado. Pero, por supuesto, dado que hay muchos vectores iniciales a los que podría aplicarse nuestra matriz, existen muchos subespacios de Krylov posibles. Solo usaremos la expresión "el subespacio de Krylov" para referirnos a un subespacio de Krylov específico ya definido para un ejemplo concreto. Para enfoques generales de resolución de problemas, nos referiremos a "un subespacio de Krylov". Una última aclaración es que también es válido hablar de un "Krylov espacio". Con frecuencia se le llama "Krylov subespacio" por su uso en el contexto de proyectar matrices de un espacio inicial a un subespacio. Siguiendo ese contexto, aquí lo llamaremos principalmente subespacio.
Comprueba tu comprensión
Explica por qué no es (a) útil, ni (b) posible, extender la dimensión del subespacio de Krylov más allá de la dimensión de la matriz de interés.
Answer
(a) Como estamos ortonormalizando los vectores a medida que los producimos, un conjunto de tales vectores formará una base completa, lo que significa que cualquier combinación lineal de ellos puede usarse para crear cualquier vector en el espacio.
(b) El proceso de ortogonalización consiste en restar la proyección de un nuevo vector sobre todos los vectores anteriores. Si todos los vectores anteriores abarcan el espacio vectorial completo, restar las proyecciones sobre el subespacio completo siempre nos dejará con un vector nulo.
Supongamos que un colega investigador está demostrando el método de Krylov aplicado a una pequeña matriz de juguete. ¿Hay algo incorrecto en su elección de la matriz y el vector inicial ?
y
Answer
Tu colega ha elegido accidentalmente un vector propio como vector inicial. Al actuar con la matriz sobre el vector inicial simplemente se devuelve el mismo vector escalado por el valor propio. Esto no generará un subespacio de dimensión creciente. Aconseja a tu colega que elija un vector inicial diferente, asegurándose de que no sea un vector propio.
Aplica el método de Krylov a la matriz dada, seleccionando un nuevo vector inicial apropiado. Escribe las estimaciones del valor propio mínimo en el orden 0 y el orden 1 de tu subespacio de Krylov.
Answer
Hay muchas respuestas posibles dependiendo de la elección del vector inicial. Elegiremos:
Para obtener aplicamos una vez a , y luego hacemos ortogonal a
En el orden 0, la proyección sobre nuestro subespacio de Krylov es
En el orden 1, la proyección sobre este subespacio de Krylov es
Esto puede hacerse a mano, pero se hace más fácilmente con numpy:
import numpy as np
vstar = np.array([[1/np.sqrt(3),1/np.sqrt(3),1/np.sqrt(3)],[-1/np.sqrt(6),np.sqrt(2/3),-1/np.sqrt(6)]]
)
A = np.array([[1, 1, 0],
[1, 1, 1],
[0, 1, 1]])
v = np.array([[1/np.sqrt(3),-1/np.sqrt(6)],[1/np.sqrt(3),np.sqrt(2/3)],[1/np.sqrt(3),-1/np.sqrt(6)]])
proj = vstar@A@v
print(proj)
eigenvalues, eigenvectors = np.linalg.eig(proj)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
outputs:
[[ 2.33333333 0.47140452]
[ 0.47140452 -0.33333333]]
The eigenvalues are [ 2.41421356 -0.41421356]
The eigenvectors are [[ 0.98559856 -0.16910198]
[ 0.16910198 0.98559856]]
La estimación del valor propio mínimo es -0,414.
1.2 Tipos de métodos de Krylov
Los "métodos de subespacios de Krylov" pueden referirse a cualquiera de varias técnicas iterativas usadas para resolver grandes sistemas lineales y problemas de valores propios. Lo que todos tienen en común es que construyen una solución aproximada a partir de un subespacio de Krylov
donde es la estimación inicial (véase la Ref [5]). Se diferencian en cómo eligen la mejor aproximación de este subespacio, equilibrando factores como la tasa de convergencia, el uso de memoria y el costo computacional total. El foco de esta lección es aprovechar la computación cuántica en el contexto de los métodos de subespacios de Krylov; una discusión exhaustiva de estos métodos queda fuera de su alcance. Las breves definiciones a continuación son solo para contexto e incluyen algunas referencias para investigar estos métodos con más profundidad.
El método del gradiente conjugado (CG): Este método se usa para resolver sistemas lineales simétricos y definidos positivos[6]. Minimiza la norma-A del error en cada iteración, haciéndolo particularmente eficaz para sistemas que surgen de EDPs elípticas discretizadas[7]. Usaremos este enfoque en la siguiente sección para motivar por qué un subespacio de Krylov sería un subespacio eficaz en el que buscar soluciones mejoradas a sistemas lineales.
El método del residuo mínimo generalizado (GMRES): Está diseñado para resolver sistemas lineales generales no simétricos. Minimiza la norma del residuo sobre un espacio de Krylov en cada iteración, haciéndolo robusto pero potencialmente intensivo en memoria para sistemas grandes[7].
El método del residuo mínimo (MINRES): Este método se usa para resolver sistemas lineales simétricos indefinidos. Es similar a GMRES pero aprovecha la simetría de la matriz para reducir el costo computacional[8].
Otros enfoques destacados incluyen el método de ortogonalización completa (FOM), estrechamente relacionado con el método de Arnoldi para problemas de valores propios, el método del gradiente bi-conjugado (BiCG) y el método de reducción de dimensión inducida (IDR).
1.3 Por qué funciona el método del subespacio de Krylov
Aquí motivaremos que el método del subespacio de Krylov debería ser una forma eficiente de aproximar los valores propios de una matriz mediante el refinamiento iterativo de las aproximaciones de los vectores propios, a través de la perspectiva del descenso más pronunciado. Argumentaremos que dada una estimación inicial del estado fundamental, el espacio de correcciones sucesivas a esa estimación inicial que produce la convergencia más rápida es un subespacio de Krylov. No llegamos hasta una demostración rigurosa del comportamiento de convergencia.
Asumamos que nuestra matriz de interés es simétrica y definida positiva. Esto hace que nuestro argumento sea más relevante para el método CG anterior. No hacemos suposiciones sobre la esparsidad aquí; ni tampoco afirmamos que deba ser hermítica (lo cual necesita ser si es un hamiltoniano).
Típicamente deseamos resolver un problema de la forma
Uno podría imaginar que donde es alguna constante, como en un problema de valores propios. Pero nuestro enunciado del problema sigue siendo más general por ahora.
Comenzamos con un vector que es una solución aproximada. Aunque hay paralelismos entre esta estimación y en la Sección 1.1, no los aprovechamos aquí. Nuestra estimación tiene un error, que llamamos
También definimos el residuo
Aquí usamos mayúscula para distinguir el residuo de la dimensión de nuestro subespacio de Krylov .

Ahora queremos dar un paso de corrección de la forma
que esperamos que mejore nuestra aproximación. Aquí es algún vector aún por determinar. Sea el error después de que se realiza la corrección. Entonces

Nos interesa cómo se comporta nuestro error cuando es transformado por nuestra matriz. Así que calculemos la norma- del error. Esto es
donde hemos usado la simetría de y también que Aquí es alguna constante independiente de . Como se menciónó en la Sección 1.2, la norma- del error no es la única cantidad que podríamos elegir minimizar, pero es una buena opción. Queremos ver cómo varía esta cantidad con nuestra elección de vectores de corrección Así que definimos la función estableciendo
es simplemente el error como función de la corrección medida en la norma-. Por lo tanto, queremos elegir de modo que sea lo más pequeño posible. Con este propósito, calculamos el gradiente de . Usando la simetría de tenemos
El gradiente apunta en la dirección del ascenso más pronunciado, lo que significa que su opuesto nos da la dirección en la que la función decrece más: la dirección del descenso más pronunciado. En nuestra estimación inicial , donde , tenemos que Así, la función decrece más en la dirección del residuo Por tanto, nuestra elección inicial se beneficiaría más de la adición del vector para algún escalar .
En el siguiente paso, elegimos de nuevo un vector y añadimos su valor a la aproximación actual. Usando el mismo argumento de antes, elegimos para algún escalar . Continuamos de esta manera, de modo que la -ésima iteración de nuestro vector es
Equivalentemente, queremos construir el espacio del que elegimos nuestras estimaciones mejoradas añadiendo , , y así sucesivamente, en orden. El -ésimo vector estimado pertenece a
Ahora, usando la relación
vemos que
Es decir, el espacio que construimos y que aproxima de forma más eficiente la solución correcta es exactamente el espacio construido mediante la aplicación sucesiva de la matriz sobre Un subespacio de Krylov es el espacio generado por los vectores de las direcciones sucesivas del descenso más pronunciado.
Por último, reiteramos que no hemos hecho ninguna afirmación numérica sobre el escalado de este enfoque, ni hemos discutido el beneficio comparativo para matrices dispersas. Esto solo pretende motivar el uso de los métodos de subespacios de Krylov y añadir cierta intuición sobre ellos. Ahora exploraremos el comportamiento de estos métodos numéricamente.
Comprueba tu comprensión
En el flujo de trabajo anterior, propusimos minimizar la norma- del error. ¿Qué otras cantidades podría uno considerar minimizar en la búsqueda del estado fundamental y su valor propio?
Answer
Uno podría imaginar usar el vector residual en lugar de la norma- del error. Puede haber casos en los que considerar el propio vector de error sea útil.
2. Métodos de Krylov en computación clásica
En esta sección implementamos iteraciones de Arnoldi computacionalmente para poder aprovechar un subespacio de Krylov en la resolución de problemas de valores propios. Primero lo aplicaremos a un ejemplo de pequeña escala y luego examinaremos cómo escala el tiempo de cómputo a medida que aumenta el tamaño de la matriz de interés. Una idea clave aquí será que la generación de los vectores que abarcan el espacio de Krylov será el mayor contribuyente al tiempo total de cómputo requerido. La memoria necesaria variará entre métodos de Krylov específicos, pero las restricciones de memoria pueden limitar el uso de los métodos de Krylov tradicionales.
2.1 Ejemplo simple de pequeña escala
En el proceso de crear un subespacio de Krylov, necesitaremos ortonormalizar los vectores de nuestro subespacio. Definamos una función que tome un vector establecido de nuestro subespacio vknown (que no se asume normalizado) y un vector candidato para agregar a nuestro subespacio vnext, y haga que vnext sea ortogonal a vknown y normalizado. Definamos además una función que recorra este proceso para todos los vectores establecidos en nuestro subespacio de Krylov para garantizar un conjunto completamente ortonormal.
# vknown is some established vector in our subspace. vnext is one we wish to add,
# which must be orthogonal to vknown.
def orthog_pair(vknown, vnext):
vknown = vknown / np.sqrt(vknown.T @ vknown)
diffvec = vknown.T @ vnext * vknown
vnext = vnext - diffvec
return vnext
# v is the candidate vector to be added to our subspace. s is the existing subspace.
def orthoset(v, s):
v = v / np.sqrt(v.T @ v)
temp = v
for i in range(len(s)):
temp = orthog_pair(s[i], temp)
v = temp / np.sqrt(temp.T @ temp)
return v
Ahora definamos una función que construya un subespacio de Krylov iterativamente cada vez más grande, hasta que el espacio de vectores de Krylov abarque el espacio completo de la matriz original. Esto nos permitirá ver qué tan bien los valores propios obtenidos con nuestro método de subespacio de Krylov coinciden con los valores exactos, en función de la dimensión del subespacio de Krylov. Cabe destacar que nuestra función krylov_full_build devuelve los vectores de Krylov, los Hamiltonianos proyectados, los valores propios y el tiempo requerido.
# Necessary imports and definitions to track time in microseconds
import time
def time_mus():
return int(time.time() * 1000000)
# This function constructs a Krylov subspace that spans the whole space of the original matrix.
# Input:
# v0 : initial vector
# matrix : original matrix to be diagonalized
# Output:
# ks : Krylov vectors
# Hs : projected Hamiltonians
# eigs : eigenvalues
# k_tot_times : time required for the operation
def krylov_full_build(v0, matrix):
t0 = time_mus()
b = v0 / np.sqrt(v0 @ v0.T)
A = matrix
ks = []
ks.append(b)
Hs = []
eigs = []
Hs.append(b.T @ A @ b)
eigs.append(np.array([b.T @ A @ b]))
k_tot_times = []
for j in range(len(A) - 1):
vec = A @ ks[j].T
ortho = orthoset(vec, ks)
ks.append(ortho)
ksarray = np.array(ks)
Hs.append(ksarray @ A @ ksarray.T)
eigs.append(np.linalg.eig(Hs[j + 1]).eigenvalues)
k_tot_times.append(time_mus() - t0)
# Return the Krylov vectors, the projected Hamiltonians, the eigenvalues,
# and the total time required.
return (ks, Hs, eigs, k_tot_times)
Probaremos esto en una matriz que aún es bastante pequeña, pero más grande de lo que quisiéramos calcular a mano.
# Define our small test matrix
test_matrix = np.array(
[
[4, -1, 0, 1, 0],
[-1, 4, -1, 2, 1],
[0, -1, 4, 3, 3],
[1, 2, 3, 4, 0],
[0, 1, 3, 0, 4],
]
)
# Give the test matrix and an initial guess as arguments in the function defined above.
# Calculate outputs.
test_ks, test_Hs, test_eigs, text_k_tot_times = krylov_full_build(
np.array([0.5, 0.5, 0, 0.5, 0.5]), test_matrix
)
Podemos verificar nuestras funciones asegurándonos de que en el último paso (cuando el espacio de Krylov es el espacio vectorial completo de la matriz original) los valores propios del método de Krylov coincidan exactamente con los de la diagonalización numérica exacta:
print(np.linalg.eig(test_matrix).eigenvalues)
print(test_eigs[len(test_matrix) - 1])
[-1.36956923 8.43756009 2.9040308 5.34436028 4.68361806]
[-1.36956923 8.43756009 2.9040308 4.68361806 5.34436028]
Eso fue exitoso. Por supuesto, lo que realmente importa es qué tan buena es nuestra aproximación en función de la dimensión de nuestro subespacio de Krylov. Como con frecuencia nos interesa encontrar estados fundamentales y otros valores propios mínimos (y por otras razones más algebraicas explicadas a continuación), veamos nuestra estimación del valor propio más bajo en función de la dimensión del subespacio de Krylov. Eso es
def errors(matrix, krylov_eigs):
targ_min = min(np.linalg.eig(matrix).eigenvalues)
err = []
for i in range(len(matrix)):
err.append(min(krylov_eigs[i]) - targ_min)
return err
import matplotlib.pyplot as plt
krylov_error = errors(test_matrix, test_eigs)
plt.plot(krylov_error)
plt.axhline(y=0, color="red", linestyle="--") # Add dashed red line at y=0
plt.xlabel("Order of Krylov subspace") # Add x-axis label
plt.ylabel("Error in minimum eigenvalue") # Add y-axis label
plt.show()
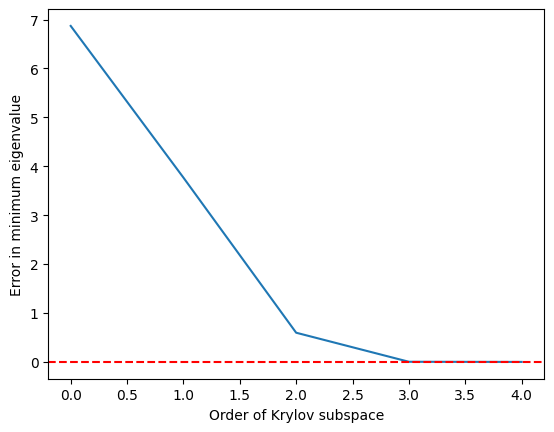
Vemos que el valor propio mínimo se alcanza con bastante precisión una vez que el subespacio de Krylov ha crecido hasta y es perfecto en
2.2 Escalado del tiempo con la dimensión de la matriz
Convenzámonos de que el método de Krylov puede ser ventajoso sobre los eigensolvers numéricos exactos de la siguiente manera:
- Construir matrices aleatorias (no dispersas, no la aplicación ideal para KQD)
- Determinar valores propios usando dos métodos: directamente con NumPy y usando un subespacio de Krylov.
- Elegimos un umbral de precisión para nuestros valores propios antes de aceptar las estimaciones de Krylov.
- Comparar el tiempo de reloj requerido para resolver de estas dos formas.
Advertencias: Como discutiremos en detalle a continuación, la diagonalización cuántica de Krylov se aplica mejor a operadores cuyas representaciones matriciales son dispersas y/o pueden escribirse usando un pequeño número de grupos de operadores de Pauli conmutativos. Las matrices aleatorias que estamos usando aquí no se ajustan a esa descripción. Solo son útiles para explorar la escala en la que los métodos clásicos de Krylov podrían ser útiles. En segundo lugar, al usar el método de Krylov calcularemos valores propios usando muchos subespacios de Krylov de diferente tamaño. Reportaremos el tiempo requerido para el subespacio de Krylov de dimensión mínima que logra la precisión requerida para el valor propio del estado fundamental. De nuevo, esto es un poco diferente a resolver un problema intratable para eigensolvers exactos, ya que estamos usando la solución exacta para evaluar la dimensión necesaria.
Comenzamos generando nuestro conjunto de matrices aleatorias.
import numpy as np
# Set the random seed
np.random.seed(42)
# how many random matrices will we make
num_matrix = 200
matrices = []
for m in range(1, num_matrix):
matrices.append(np.random.rand(m, m))
Ahora diagonalizamos cada matriz directamente, usando numpy. Calculamos el tiempo requerido para la diagonalización para su comparación posterior.
matrix_numpy_times = []
matrix_numpy_eigs = []
for mm in range(num_matrix - 1):
t0 = time_mus()
matrix_numpy_eigs.append(min(np.linalg.eig(matrices[mm]).eigenvalues))
matrix_numpy_times.append(time_mus() - t0)
plt.plot(matrix_numpy_times)
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()
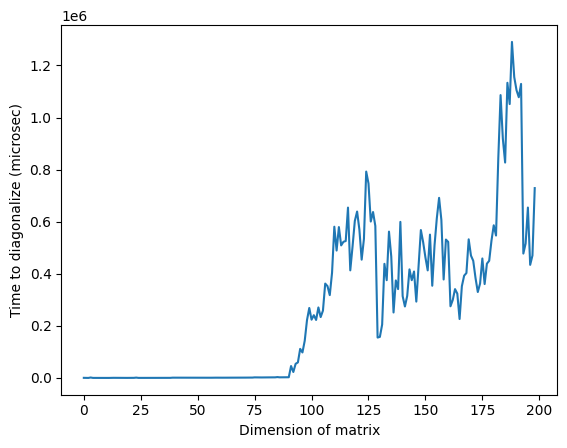
Ten en cuenta que en la imagen anterior, el tiempo anómalamente alto alrededor de la dimensión 125 puede deberse a la naturaleza aleatoria de las matrices o a la implementación en el procesador clásico utilizado, pero no es reproducible. Al volver a ejecutar el código se obtendrá un perfil diferente con distintos picos anómalos.
Ahora, para cada matriz construiremos un subespacio de Krylov y calcularemos valores propios en pasos. En cada paso, verificaremos si el valor propio más bajo se ha obtenido dentro de nuestro error absoluto especificado. El subespacio que primero nos dé valores propios dentro del error especificado es el subespacio para el que registraremos los tiempos de cómputo. Ejecutar esta celda puede tardar varios minutos, dependiendo de la velocidad del procesador. Puedes omitir la evaluación o reducir la dimensión máxima de las matrices diagonalizadas. Revisar los resultados precalculados es suficiente.
# Choose the absolute error you can tolerate, and make a list for tracking the Krylov subspace size
# at which that error is achieved.
abserr = 0.05
accept_subspace_size = []
# Lists to store total time spent on the Krylov method, and the subset of that time spent on
# diagonalizing the projected matrix.
matrix_krylov_tot_times = []
matrix_krylov_dim = []
# Step through all our random matrices
for mm in range(0, num_matrix - 1):
test_ks, test_Hs, test_eigs, test_k_tot_times = krylov_full_build(
np.ones(len(matrices[mm])), matrices[mm]
)
# We have not yet found a Krylov subspace that produces our minimum eigenvalue to
# within the required error.
found = 0
for j in range(0, len(matrices[mm]) - 1):
# If we still haven't found the desired subspace...
if found == 0:
# ...but if this one satisfies the requirement, then record everything
if (
abs((min(test_eigs[j]) - matrix_numpy_eigs[mm]) / matrix_numpy_eigs[mm])
< abserr
):
accept_subspace_size.append(j)
matrix_krylov_tot_times.append(test_k_tot_times[j])
matrix_krylov_dim.append(mm)
found = 1
Grafiquemos los tiempos que hemos obtenido para estos dos métodos a modo de comparación:
plt.plot(matrix_numpy_times, color="blue")
plt.plot(matrix_krylov_dim, matrix_krylov_tot_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()

Estos son los tiempos reales requeridos, pero para fines de discusión, suavicemos estas curvas promediando sobre algunos puntos/dimensiones de matrices adyacentes. Esto se hace a continuación:
smooth_numpy_times = []
smooth_krylov_times = []
# Choose the number of adjacent points over which to average forward;
# the same will be used backward.
smooth_steps = 10
# We will do this smoothing for all points/matrix dimensions
for i in range(len(matrix_krylov_tot_times)):
# Ensure we don't exceed the boundaries of our lists
start = max(0, i - smooth_steps)
end = min(len(matrix_krylov_tot_times) - 1, i + smooth_steps)
# Dummy variables for accumulating an average over adjacent points. This is done for both Krylov
# and the NumPy calculations.
smooth_count = 0
smooth_numpy_sum = 0
smooth_krylov_sum = 0
for j in range(start, end):
smooth_numpy_sum = smooth_numpy_sum + matrix_numpy_times[j]
smooth_krylov_sum = smooth_krylov_sum + matrix_krylov_tot_times[j]
smooth_count = smooth_count + 1
# Appending the averaged adjacent values to our new smooth lists
smooth_numpy_times.append(smooth_numpy_sum / smooth_count)
smooth_krylov_times.append(smooth_krylov_sum / smooth_count)
plt.plot(smooth_numpy_times, color="blue")
plt.plot(smooth_krylov_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (smoothed, microsec)") # Add y-axis label
plt.show()

Ten en cuenta que el tiempo requerido para construir un subespacio de Krylov inicialmente supera el tiempo requerido para la diagonalización completa de numpy. Pero a medida que crece el tamaño de la matriz, el método de Krylov se vuelve ventajoso. Esto es cierto incluso si reducimos nuestro error aceptable, aunque la ventaja aparece a un tamaño de matriz mayor. Vale la pena analizar esto.
La complejidad temporal de la diagonalización numérica es (con alguna variación entre algoritmos). La complejidad temporal de generar una base ortonormal de vectores también es . Por lo tanto, la ventaja del método de Krylov no está relacionada con el uso de base ortonormal, sino con el uso de una base ortonormal particular que selecciona eficazmente los valores propios de interés. Ya vimos esto en el bosquejo de una demostración en la primera sección de esta lección, y esto es fundamental para las garantías de convergencia en los métodos de Krylov.
Revisemos nuestro progreso hasta ahora:
- Para matrices muy grandes, el método del subespacio de Krylov puede producir valores propios aproximados dentro de las tolerancias requeridas más rápido que los algoritmos de diagonalización tradicionales.
- Para tales matrices muy grandes, la generación de un subespacio de Krylov es la parte más costosa en tiempo del método del subespacio de Krylov.
- Por lo tanto, una forma eficiente de generar un subespacio de Krylov sería muy valiosa.
Aquí es finalmente donde entra en escena la computadora cuántica.
Comprueba tu comprensión
Consulta la gráfica suavizada de tiempos de diagonalización frente a la dimensión de la matriz que se muestra arriba.
(a) ¿Aproximadamente en qué dimensión de la matriz se volvió más rápido el método de Krylov, según esta gráfica?
(b) ¿Qué aspectos del cálculo podrían cambiar la dimensión a la que el método de Krylov se vuelve más rápido?
Answer
(a) Las respuestas pueden variar si vuelves a ejecutar el cálculo, pero el método de Krylov se vuelve más rápido aproximadamente en la dimensión 80–85.
(b) Hay muchas respuestas posibles. Algunos factores importantes son la precisión que requerimos y la dispersión (sparsity) de las matrices que se están diagonalizando.
3. Krylov mediante evolución temporal
Todo lo que hemos descrito hasta ahora puede realizarse de forma clásica. ¿Cómo y cuándo usaríamos entonces un ordenador cuántico? Para matrices muy grandes, el método de Krylov puede requerir tiempos de cómputo largos y grandes cantidades de memoria. El tiempo necesario para la operación matricial de sobre escala como en el peor caso. Incluso multiplicar matrices dispersas por un vector (el caso típico en los solvers clásicos de tipo Krylov) tiene una complejidad temporal que escala como . Esto se realiza para cada vector que queremos incluir en nuestro subespacio. La dimensión del subespacio generalmente no es una fracción significativa de , y a menudo escala como . Por tanto, generar todos los vectores escala como en el peor caso. Aunque hay otros pasos, como la ortonormalización, este es el escalado dominante que hay que tener en mente.
La computación cuántica nos permite cambiar qué atributos del problema determinan el escalado del tiempo y los recursos necesarios. En lugar de depender del tamaño de la matriz en general, veremos cosas como el número de shots y el número de términos de Pauli no conmutativos que componen el Hamiltoniano. Exploremos cómo funciona esto.
3.1 Evolución temporal
Recuerda que el operador que hace evolucionar temporalmente un estado cuántico es (y es muy común, especialmente en computación cuántica, omitir el de la notación). Una forma de entender e incluso realizar dicha función exponencial de un operador es observar su expansión en series de Taylor. Nótese que esta operación actuando sobre algún vector inicial produce una suma de términos con potencias crecientes de aplicadas al estado inicial. ¡Parece que podemos construir nuestro subespacio de Krylov simplemente haciendo evolucionar temporalmente nuestro estado de suposición inicial!
La dificultad está en realizar la evolución temporal en un ordenador cuántico real. Muchos de los términos en el Hamiltoniano no conmutarán entre sí. Así que, aunque algunos operadores exponenciales simples como corresponden a circuitos sencillos, los Hamiltonianos generales no. Y dado que contienen términos no conmutativos, no podemos simplemente descomponer el exponencial en un producto de exponenciales simples, como sí podemos hacer con números.
Esto no es trivial, pero es un proceso bien estudiado en computación cuántica. Llevamos a cabo la evolución temporal en ordenadores cuánticos mediante un proceso llamado trotterización, que en sí mismo es un tema rico[10]. Pero a muy alto nivel, al dividir la evolución temporal en pasos muy pequeños, digamos pasos de tamaño , limitamos los efectos de la no conmutatividad de los términos.
donde .
Llamemos "subespacio de Krylov de potencias" al subespacio de Krylov de orden r que generamos en el contexto clásico usando directamente potencias de H.
Ahora generamos un espacio similar usando el operador unitario de evolución temporal ; nos referiremos a esto como el "espacio de Krylov unitario" . El subespacio de Krylov de potencias que usamos clásicamente no puede generarse directamente en un ordenador cuántico, ya que no es un operador unitario. Se puede demostrar que usar el subespacio de Krylov unitario proporciona garantías de convergencia similares a las del subespacio de Krylov de potencias; concretamente, el error del estado fundamental converge eficientemente siempre que el estado inicial tenga un solapamiento con el verdadero estado fundamental que no sea exponencialmente pequeño, y siempre que exista una brecha suficiente entre autovalores. Véase la Ref [1] para una discusión más precisa de la convergencia.
Aquí, las potencias de se convierten en diferentes pasos temporales (la potencia de avanza un tiempo ). Podemos etiquetar el elemento del subespacio que ha evolucionado temporalmente por un tiempo total como .
Podemos proyectar nuestro Hamiltoniano H sobre el subespacio de Krylov unitario, . En otras palabras, calculamos cada elemento matricial de en la base . Nos referiremos a esta matriz proyectada como .
3.2 Cómo implementarlo en un ordenador cuántico
Los elementos matriciales de vienen dados por los valores esperados , que pueden estimarse usando el ordenador cuántico. Ten en cuenta que puede escribirse como una suma de operadores de Pauli sobre diferentes qubits, y que no todos los operadores de Pauli pueden medirse simultáneamente. Podemos ordenar los términos de Pauli en grupos de términos conmutativos y medir todos a la vez. Pero puede que necesitemos muchos de estos grupos para cubrir todos los términos. Por ello, el número de grupos conmutativos distintos en que los términos pueden particionarse, , se vuelve importante.
Aquí es una cadena de Pauli de la forma o un conjunto de tales cadenas de Pauli que conmutan entre sí. Dado que podemos escribir como dicha suma de operadores medibles, las siguientes expresiones para los elementos matriciales de pueden realizarse usando el primitivo Estimator de Qiskit Runtime.
Donde son los vectores del espacio de Krylov unitario y son los múltiplos del paso temporal elegido. En un ordenador cuántico, el cálculo de cada elemento matricial puede realizarse con cualquier algoritmo que nos permita obtener el solapamiento entre estados cuánticos. En esta lección nos centraremos en el test de Hadamard. Dado que tiene dimensión , el Hamiltoniano proyectado en el subespacio tendrá dimensiones . Con suficientemente pequeño (generalmente es suficiente para obtener convergencia de las estimaciones de autovalores) podemos entonces diagonalizar clásicamente el Hamiltoniano proyectado . Sin embargo, no podemos diagonalizar directamente debido a la no ortogonalidad de los vectores del espacio de Krylov. Tendremos que medir sus solapamientos y construir una matriz
Esto nos permite resolver el problema de autovalores en un espacio no ortogonal (también llamado problema de autovalores generalizado)
A continuación se pueden obtener estimaciones de los autovalores y autoestados de observando las soluciones de este problema de autovalores generalizado. Por ejemplo, la estimación de la energía del estado fundamental se obtiene tomando el autovalor más pequeño y el estado fundamental del autovector correspondiente . Los coeficientes en determinan la contribución de los diferentes vectores que generan .
Problema de autovalores generalizado
¿Por qué no podemos simplemente diagonalizar ? Dado que contiene información sobre la geometría de la base de Krylov (que es no ortogonal en todos los casos salvo los muy especiales), por sí sola no describe una proyección del Hamiltoniano completo, por lo que sus autovalores no tienen ninguna relación particular con los del Hamiltoniano completo — podrían ser cualquier valor aleatorio. Resolver el problema de autovalores generalizado es necesario para obtener los autovalores y autovectores aproximados correspondientes a la proyección del Hamiltoniano completo en el espacio de Krylov.
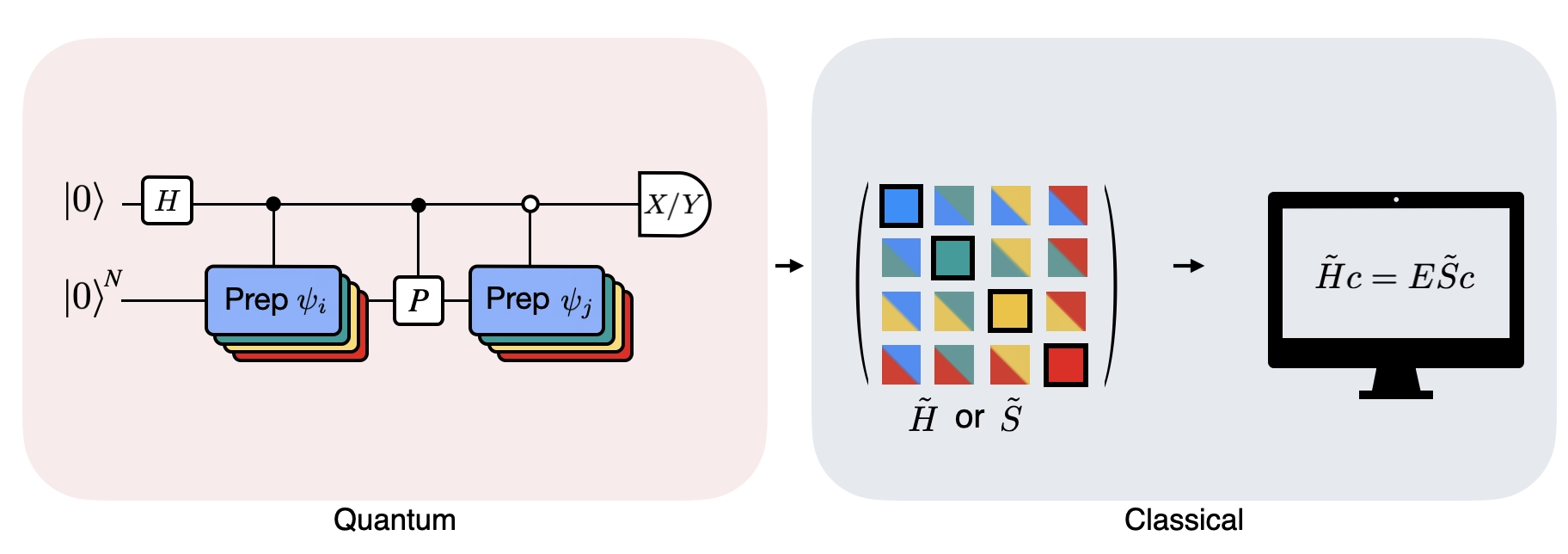
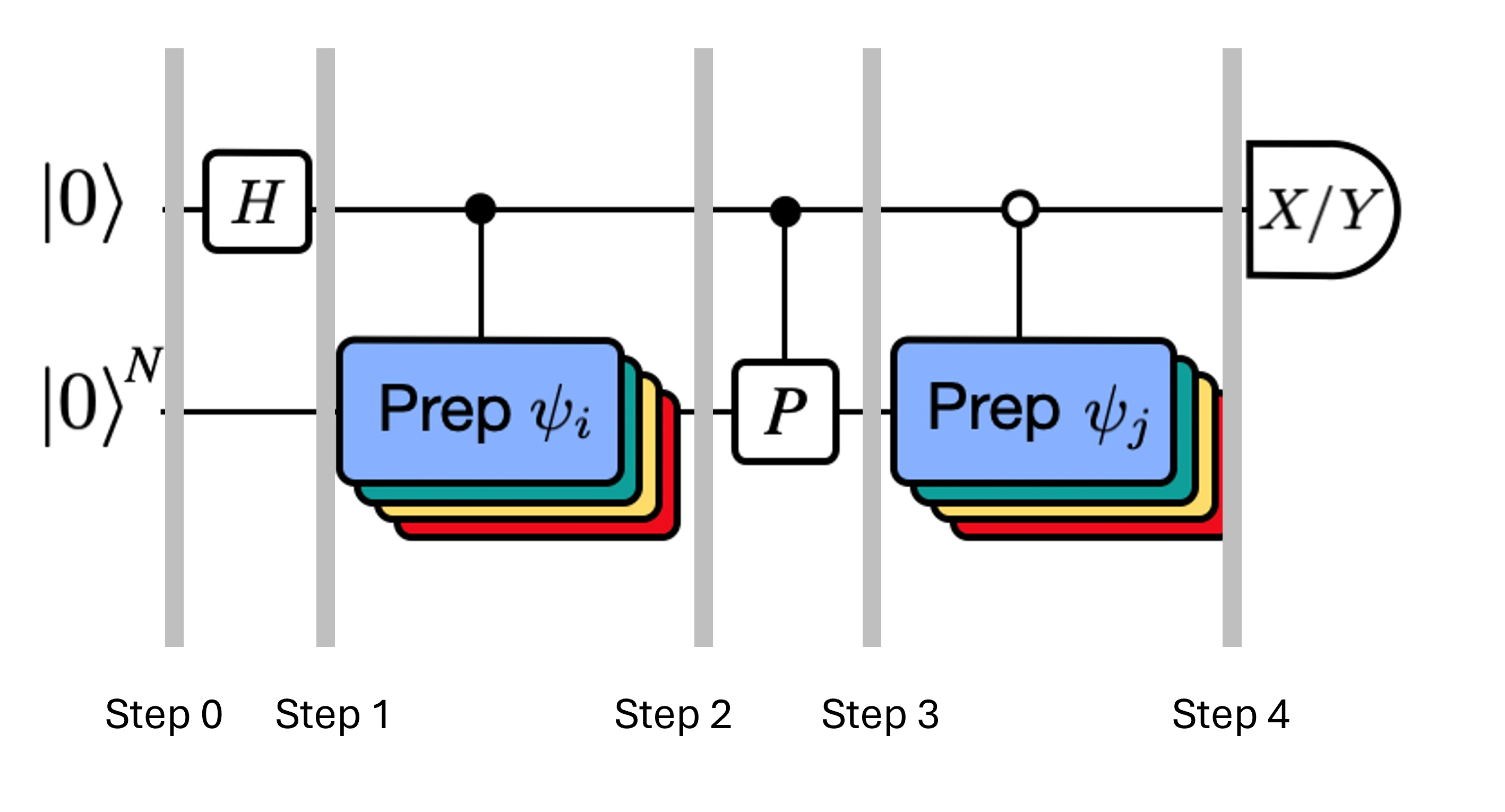
La figura muestra una representación en circuito del test de Hadamard modificado, un método que se utiliza para calcular el solapamiento entre diferentes estados cuánticos. Para cada elemento matricial , se realiza un test de Hadamard entre los estados , . Esto se resalta en la figura mediante el esquema de colores para los elementos matriciales y las correspondientes operaciones , . Así, para calcular todos los elementos matriciales del Hamiltoniano proyectado se requiere un conjunto de tests de Hadamard para todas las combinaciones posibles de vectores del espacio de Krylov. El hilo superior en el circuito del test de Hadamard es un qubit ancilla que se mide en la base X o Y; su valor esperado determina el valor del solapamiento entre los estados. El hilo inferior representa todos los qubits del Hamiltoniano del sistema. La operación prepara el qubit del sistema en el estado controlado por el estado del qubit ancilla (de manera similar para ) y la operación representa la descomposición de Pauli del Hamiltoniano del sistema . La implementación de esto en un ordenador cuántico se muestra con más detalle a continuación.
4. Diagonalización cuántica de Krylov en un ordenador cuántico
Ahora implementaremos la diagonalización cuántica de Krylov en un ordenador cuántico real. Empecemos importando algunos paquetes útiles.
import numpy as np
import scipy as sp
import matplotlib.pylab as plt
from typing import Union, List
import warnings
from qiskit.quantum_info import SparsePauliOp, Pauli
from qiskit.circuit import Parameter
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit.library import PauliEvolutionGate
from qiskit.synthesis import LieTrotter
# from qiskit.providers.fake_provider import Fake20QV1
from qiskit_ibm_runtime import QiskitRuntimeService, EstimatorV2 as Estimator, Batch
import itertools as it
warnings.filterwarnings("ignore")
A continuación definimos la función para resolver el problema de autovalores generalizado que acabamos de explicar.
def solve_regularized_gen_eig(
h: np.ndarray,
s: np.ndarray,
threshold: float,
k: int = 1,
return_dimn: bool = False,
) -> Union[float, List[float]]:
"""
Method for solving the generalized eigenvalue problem with regularization
Args:
h (numpy.ndarray):
The effective representation of the matrix in our Krylov subspace
s (numpy.ndarray):
The matrix of overlaps between vectors of our Krylov subspace
threshold (float):
Cut-off value for the eigenvalue of s
k (int):
Number of eigenvalues to return
return_dimn (bool):
Whether to return the size of the regularized subspace
Returns:
lowest k-eigenvalue(s) that are the solution of the regularized generalized eigenvalue problem
"""
s_vals, s_vecs = sp.linalg.eigh(s)
s_vecs = s_vecs.T
good_vecs = np.array([vec for val, vec in zip(s_vals, s_vecs) if val > threshold])
h_reg = good_vecs.conj() @ h @ good_vecs.T
s_reg = good_vecs.conj() @ s @ good_vecs.T
if k == 1:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][0], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][0]
else:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][:k], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][:k]
Al menos en los benchmarks iniciales, es útil conocer una solución clásica exacta para verificar el comportamiento de convergencia. La función a continuación calcula la energía del estado fundamental de un Hamiltoniano, usando el Hamiltoniano y el número de qubits como argumentos.
def single_particle_gs(H_op, n_qubits):
"""
Find the ground state of the single particle(excitation) sector
"""
H_x = []
for p, coeff in H_op.to_list():
H_x.append(set([i for i, v in enumerate(Pauli(p).x) if v]))
H_z = []
for p, coeff in H_op.to_list():
H_z.append(set([i for i, v in enumerate(Pauli(p).z) if v]))
H_c = H_op.coeffs
print("n_sys_qubits", n_qubits)
n_exc = 1
sub_dimn = int(sp.special.comb(n_qubits + 1, n_exc))
print("n_exc", n_exc, ", subspace dimension", sub_dimn)
few_particle_H = np.zeros((sub_dimn, sub_dimn), dtype=complex)
sparse_vecs = [
set(vec) for vec in it.combinations(range(n_qubits + 1), r=n_exc)
] # list all of the possible sets of n_exc indices of 1s in n_exc-particle states
m = 0
for i, i_set in enumerate(sparse_vecs):
for j, j_set in enumerate(sparse_vecs):
m += 1
if len(i_set.symmetric_difference(j_set)) <= 2:
for p_x, p_z, coeff in zip(H_x, H_z, H_c):
if i_set.symmetric_difference(j_set) == p_x:
sgn = ((-1j) ** len(p_x.intersection(p_z))) * (
(-1) ** len(i_set.intersection(p_z))
)
else:
sgn = 0
few_particle_H[i, j] += sgn * coeff
gs_en = min(np.linalg.eigvalsh(few_particle_H))
print("single particle ground state energy: ", gs_en)
return gs_en
4.1 Paso 1: Mapear el problema a circuitos y operadores cuánticos
Ahora definiremos un Hamiltoniano. Esto es diferente de la función anterior en que esa función toma un Hamiltoniano como argumento y devuelve solo el estado fundamental, y lo hace clásicamente. Este Hamiltoniano que definimos aquí determina los niveles de energía de todos los autoestados de energía, y puede construirse usando operadores de Pauli e implementarse en un ordenador cuántico.
Elegimos un Hamiltoniano correspondiente a una cadena de espines que pueden tener cualquier orientación en el espacio, llamada "cadena de Heisenberg". Asumimos que el espín puede ser influenciado por sus vecinos más cercanos (los espines e ) pero no por vecinos más distantes. También permitimos la posibilidad de que la interacción entre espines sea diferente cuando los espines apuntan a lo largo de diferentes ejes. Esta asimetría surge a veces, por ejemplo, debido a la estructura de la red cristalina en la que están inmersos los espines.
# Define problem Hamiltonian.
n_qubits = 10
# coupling strength for XX, YY, and ZZ interactions
JX = 1
JY = 3
JZ = 2
# Define the Hamiltonian:
H_int = [["I"] * n_qubits for _ in range(3 * (n_qubits - 1))]
for i in range(n_qubits - 1):
H_int[i][i] = "Z"
H_int[i][i + 1] = "Z"
for i in range(n_qubits - 1):
H_int[n_qubits - 1 + i][i] = "X"
H_int[n_qubits - 1 + i][i + 1] = "X"
for i in range(n_qubits - 1):
H_int[2 * (n_qubits - 1) + i][i] = "Y"
H_int[2 * (n_qubits - 1) + i][i + 1] = "Y"
H_int = ["".join(term) for term in H_int]
H_tot = [
(term, JZ)
if term.count("Z") == 2
else (term, JY)
if term.count("Y") == 2
else (term, JX)
for term in H_int
]
# Get operator
H_op = SparsePauliOp.from_list(H_tot)
print(H_tot)
[('ZZIIIIIIII', 2), ('IZZIIIIIII', 2), ('IIZZIIIIII', 2), ('IIIZZIIIII', 2), ('IIIIZZIIII', 2), ('IIIIIZZIII', 2), ('IIIIIIZZII', 2), ('IIIIIIIZZI', 2), ('IIIIIIIIZZ', 2), ('XXIIIIIIII', 1), ('IXXIIIIIII', 1), ('IIXXIIIIII', 1), ('IIIXXIIIII', 1), ('IIIIXXIIII', 1), ('IIIIIXXIII', 1), ('IIIIIIXXII', 1), ('IIIIIIIXXI', 1), ('IIIIIIIIXX', 1), ('YYIIIIIIII', 3), ('IYYIIIIIII', 3), ('IIYYIIIIII', 3), ('IIIYYIIIII', 3), ('IIIIYYIIII', 3), ('IIIIIYYIII', 3), ('IIIIIIYYII', 3), ('IIIIIIIYYI', 3), ('IIIIIIIIYY', 3)]
El código a continuación restringe el Hamiltoniano a estados de una sola partícula y usa la norma espectral para establecer un buen tamaño para nuestro paso temporal . Elegimos heurísticamente un valor para el paso temporal dt (basado en cotas superiores de la norma del Hamiltoniano). La Ref [9] demostró que un paso temporal suficientemente pequeño es , y que es preferible hasta cierto punto subestimar este valor en lugar de sobreestimarlo, ya que sobreestimarlo puede permitir que las contribuciones de estados de alta energía corrompan incluso el estado óptimo en el espacio de Krylov. Por otro lado, elegir demasiado pequeño lleva a un peor condicionamiento del subespacio de Krylov, ya que los vectores de la base de Krylov difieren menos de un paso temporal al siguiente.
# Get Hamiltonian restricted to single-particle states
single_particle_H = np.zeros((n_qubits, n_qubits))
for i in range(n_qubits):
for j in range(i + 1):
for p, coeff in H_op.to_list():
p_x = Pauli(p).x
p_z = Pauli(p).z
if all(p_x[k] == ((i == k) + (j == k)) % 2 for k in range(n_qubits)):
sgn = ((-1j) ** sum(p_z[k] and p_x[k] for k in range(n_qubits))) * (
(-1) ** p_z[i]
)
else:
sgn = 0
single_particle_H[i, j] += sgn * coeff
for i in range(n_qubits):
for j in range(i + 1, n_qubits):
single_particle_H[i, j] = np.conj(single_particle_H[j, i])
# Set dt according to spectral norm
dt = np.pi / np.linalg.norm(single_particle_H, ord=2)
dt
np.float64(0.17453292519943295)
Especificamos el número de pasos de Trotter a usar en la evolución temporal. También especificamos una dimensión máxima de Krylov de 4. Esta dimensión de Krylov no es suficientemente grande para aplicaciones realistas. Pero es suficiente para este ejemplo. Además, verificaremos la convergencia con dimensiones aún menores. Exploraremos métodos en lecciones posteriores que nos permiten escalar y proyectar nuestros Hamiltonianos sobre subespacios más grandes.
# Set parameters for quantum Krylov algorithm
krylov_dim = 4 # size of krylov subspace
num_trotter_steps = 4
dt_circ = dt / num_trotter_steps
Preparación del estado
Elige un estado de referencia que tenga algún solapamiento con el estado fundamental. Para este Hamiltoniano, usamos un estado con una excitación en el qubit central como nuestro estado de referencia.
qc_state_prep = QuantumCircuit(n_qubits)
qc_state_prep.x(int(n_qubits / 2) + 1)
qc_state_prep.draw("mpl", scale=0.5)

Evolución temporal
Podemos realizar el operador de evolución temporal generado por un Hamiltoniano dado: mediante la aproximación de Lie-Trotter. Por simplicidad usamos el PauliEvolutionGate integrado en el circuito de evolución temporal. La sintaxis general para esto es la siguiente.
t = Parameter("t")
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=t, synthesis=LieTrotter(reps=num_trotter_steps)
)
qr = QuantumRegister(n_qubits)
qc_evol = QuantumCircuit(qr)
qc_evol.append(evol_gate, qargs=qr)
<qiskit.circuit.instructionset.InstructionSet at 0x7ccaa4664250>
Utilizaremos una versión de esto más adelante en el test de Hadamard, avanzando en el tiempo en pasos de .
Test de Hadamard
Recuerda que queremos calcular los elementos de matriz tanto de como de la matriz de Gram usando el test de Hadamard. Repasemos cómo funciona esto en este contexto, centrándonos primero en la construcción de El proceso general se representa gráficamente a continuación. Las capas de bloques de preparación de estado de colores sirven como recordatorio de que este proceso se lleva a cabo para todas las combinaciones de y de nuestro subespacio.
Los estados del sistema en los pasos indicados son:
Aquí es un término de Pauli en la descomposición del Hamiltoniano (nota que no puede ser una combinación lineal de múltiples términos de Pauli que conmuten, ya que eso no sería unitario — el agrupamiento es posible usando una construcción diferente que mostraremos más adelante). , son operaciones controladas que preparan los vectores , del espacio de Krylov unitario, con . Aplicar mediciones de e a este circuito calcula las partes real e imaginaria, respectivamente, de los elementos de matriz que necesitamos.
Partiendo del Paso 4 anterior, aplica la puerta de Hadamard al qubit cero.
Luego mide o .
A partir de la identidad . De manera similar, medir da
Añadiendo estos pasos a la evolución temporal que configuramos anteriormente, escribimos lo siguiente.
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
## Create the time-evo op dagger circuit
evol_gate_d = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
evol_gate_d = evol_gate_d.inverse()
# Put pieces together
qc_reg = QuantumRegister(n_qubits)
qc_temp = QuantumCircuit(qc_reg)
qc_temp.compose(qc_state_prep, inplace=True)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate, qargs=qc_reg)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate_d, qargs=qc_reg)
qc_temp.compose(qc_state_prep.inverse(), inplace=True)
# Create controlled version of the circuit
controlled_U = qc_temp.to_gate().control(1)
# Create hadamard test circuit for real part
qr = QuantumRegister(n_qubits + 1)
qc_real = QuantumCircuit(qr)
qc_real.h(0)
qc_real.append(controlled_U, list(range(n_qubits + 1)))
qc_real.h(0)
print("Circuit for calculating the real part of the overlap in S via Hadamard test")
qc_real.draw("mpl", fold=-1, scale=0.5)
Circuit for calculating the real part of the overlap in S via Hadamard test

Ya advertimos sobre la profundidad de los circuitos de Trotter. Realizar el test de Hadamard en estas condiciones puede producir un circuito aún más profundo, especialmente una vez que lo descomponemos en puertas nativas. Esto aumentará aún más si tenemos en cuenta la topología del dispositivo. Por eso, antes de usar tiempo en el ordenador cuántico, es una buena idea verificar la profundidad de puertas de 2 qubits de nuestro circuito.
print(
"Number of layers of 2Q operations",
qc_real.decompose(reps=2).depth(lambda x: x[0].num_qubits == 2),
)
Number of layers of 2Q operations 14401
Un circuito de esta profundidad no puede devolver resultados utilizables en los ordenadores cuánticos actuales. Si queremos construir y necesitamos un método mejor. Esta es la razón del test de Hadamard eficiente que se presenta a continuación.
4. 2 Paso 2. Optimizar circuitos y operadores para el hardware objetivo
Test de Hadamard eficiente
Podemos optimizar los circuitos profundos para el test de Hadamard que hemos obtenido introduciendo algunas aproximaciones y apoyándonos en ciertas suposiciones sobre el Hamiltoniano del modelo. Por ejemplo, considera el siguiente circuito para el test de Hadamard:
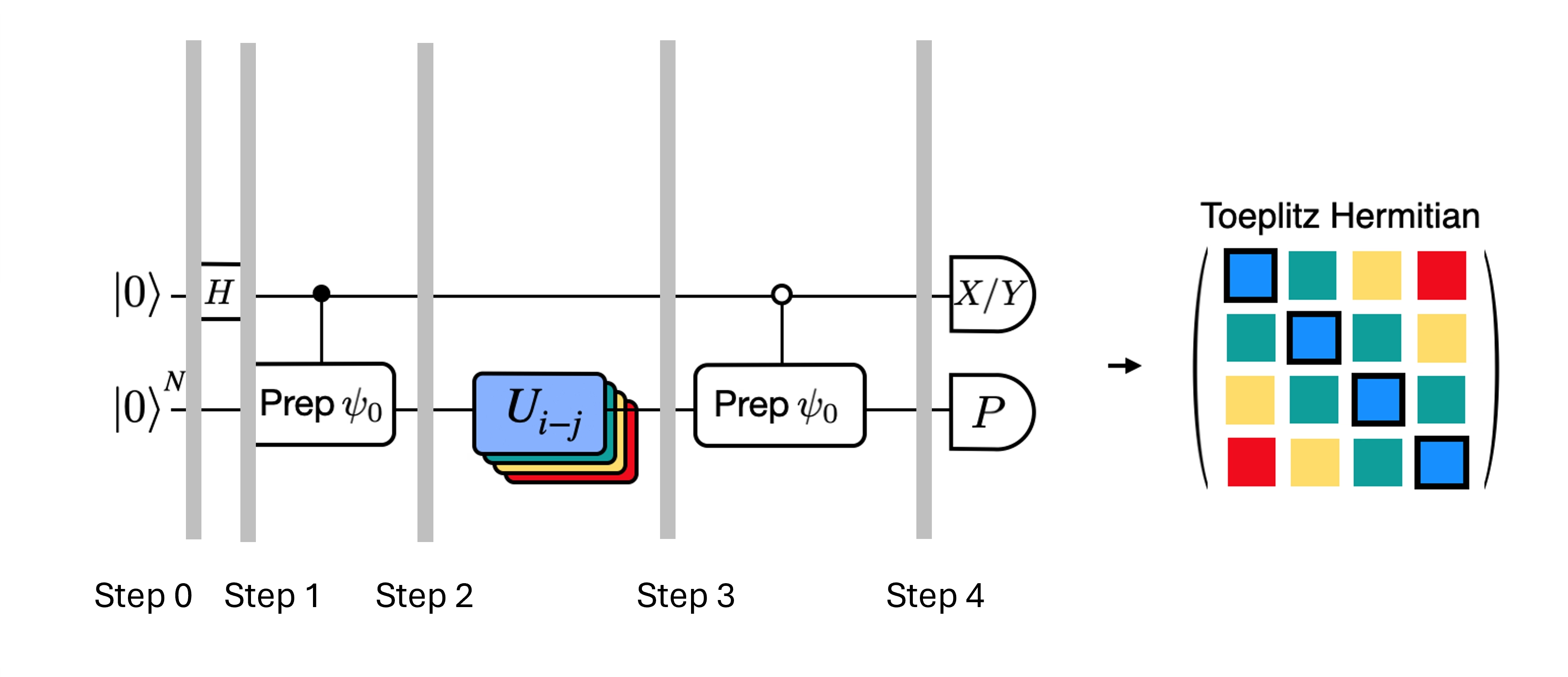
Supón que podemos calcular clásicamente , el valor propio de bajo el Hamiltoniano . Esto se satisface cuando el Hamiltoniano preserva la simetría U(1). Aunque esto puede parecer una suposición fuerte, hay muchos casos en que es razonable asumir que existe un estado de vacío (que en este caso se corresponde con el estado ) que no se ve afectado por la acción del Hamiltoniano. Esto es cierto, por ejemplo, para los Hamiltonianos de química que describen moléculas estables (donde el número de electrones se conserva). Dado que la puerta prepara el estado de referencia deseado , por ejemplo, para preparar el estado HF en química sería un producto de NOTs de un solo qubit, por lo que controlado es simplemente un producto de CNOTs. Entonces el circuito anterior implementa el siguiente estado antes de la medición:
donde hemos usado el desplazamiento de fase calculable clásicamente del paso 2 al 3. Por lo tanto, los valores esperados son
Usando estas suposiciones pudimos escribir los valores esperados de los operadores de interés con menos operaciones controladas. De hecho, solo necesitamos implementar la preparación de estado controlada y no las evoluciones temporales controladas. Reformular nuestro cálculo como se indica arriba nos permitirá reducir significativamente la profundidad de los circuitos resultantes.
Ten en cuenta que, como beneficio adicional, dado que el operador de Pauli aparece ahora como una medición al final del circuito en lugar de como una puerta controlada en el centro, puede medirse junto con otros operadores de Pauli que conmuten, como en la descomposición indicada anteriormente.
Descomponer el operador de evolución temporal con la descomposición de Trotter
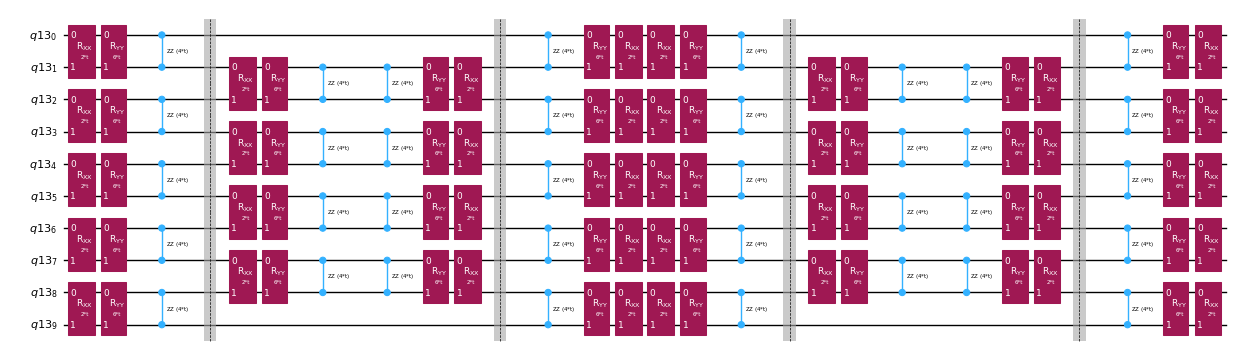
En lugar de implementar el operador de evolución temporal de forma exacta, podemos usar la descomposición de Trotter para implementar una aproximación. Repetir varias veces una descomposición de Trotter de cierto orden nos permite reducir aún más el error introducido por la aproximación. A continuación, construimos directamente la implementación de Trotter de la manera más eficiente para el grafo de interacciones del Hamiltoniano que estamos considerando (solo interacciones de primeros vecinos). En la práctica, insertamos rotaciones de Pauli , , con constantes de acoplamiento y y un ángulo parametrizado , que corresponden a la implementación aproximada de . Dada la diferencia en la definición de las rotaciones de Pauli y la evolución temporal que queremos implementar, tendremos que usar el parámetro para lograr una evolución temporal de . Además, invertimos el orden de las operaciones para los números impares de repeticiones de los pasos de Trotter, lo que es funcionalmente equivalente pero permite sintetizar operaciones adyacentes en una única unitaria . Esto produce un circuito mucho más superficial que el obtenido con la funcionalidad genérica PauliEvolutionGate().
t = Parameter("t")
# Create instruction for rotation about XX+YY-ZZ:
Rxyz_circ = QuantumCircuit(2)
Rxyz_circ.rxx(2 * JX * t, 0, 1)
Rxyz_circ.ryy(2 * JY * t, 0, 1)
Rxyz_circ.rzz(2 * JZ * t, 0, 1)
Rxyz_instr = Rxyz_circ.to_instruction(label="R J_x XX + J_y YY + J_z ZZ")
interaction_list = [
[[i, i + 1] for i in range(0, n_qubits - 1, 2)],
[[i, i + 1] for i in range(1, n_qubits - 1, 2)],
] # linear chain
qr = QuantumRegister(n_qubits)
trotter_step_circ = QuantumCircuit(qr)
for i, color in enumerate(interaction_list):
for interaction in color:
trotter_step_circ.append(Rxyz_instr, interaction)
if i < len(interaction_list) - 1:
trotter_step_circ.barrier()
reverse_trotter_step_circ = trotter_step_circ.reverse_ops()
qc_evol = QuantumCircuit(qr)
for step in range(num_trotter_steps):
if step % 2 == 0:
qc_evol = qc_evol.compose(trotter_step_circ)
else:
qc_evol = qc_evol.compose(reverse_trotter_step_circ)
qc_evol.decompose().draw("mpl", fold=-1, scale=0.5)
Preparamos un estado inicial nuevamente para este test de Hadamard eficiente.
control = 0
excitation = int(n_qubits / 2) + 1
controlled_state_prep = QuantumCircuit(n_qubits + 1)
controlled_state_prep.cx(control, excitation)
controlled_state_prep.draw("mpl", fold=-1, scale=0.5)

Circuitos plantilla para calcular los elementos de matriz de y mediante el test de Hadamard
La única diferencia entre los circuitos usados en el test de Hadamard será la fase en el operador de evolución temporal y los observables medidos. Por lo tanto, podemos preparar un circuito plantilla que represente el circuito genérico para el test de Hadamard, con marcadores de posición para las puertas que dependen del operador de evolución temporal.
# Parameters for the template circuits
parameters = []
for idx in range(1, krylov_dim):
parameters.append(dt_circ * (idx))
# Create modified hadamard test circuit
qr = QuantumRegister(n_qubits + 1)
qc = QuantumCircuit(qr)
qc.h(0)
qc.compose(controlled_state_prep, list(range(n_qubits + 1)), inplace=True)
qc.barrier()
qc.compose(qc_evol, list(range(1, n_qubits + 1)), inplace=True)
qc.barrier()
qc.x(0)
qc.compose(controlled_state_prep.inverse(), list(range(n_qubits + 1)), inplace=True)
qc.x(0)
qc.decompose().draw("mpl", fold=-1)
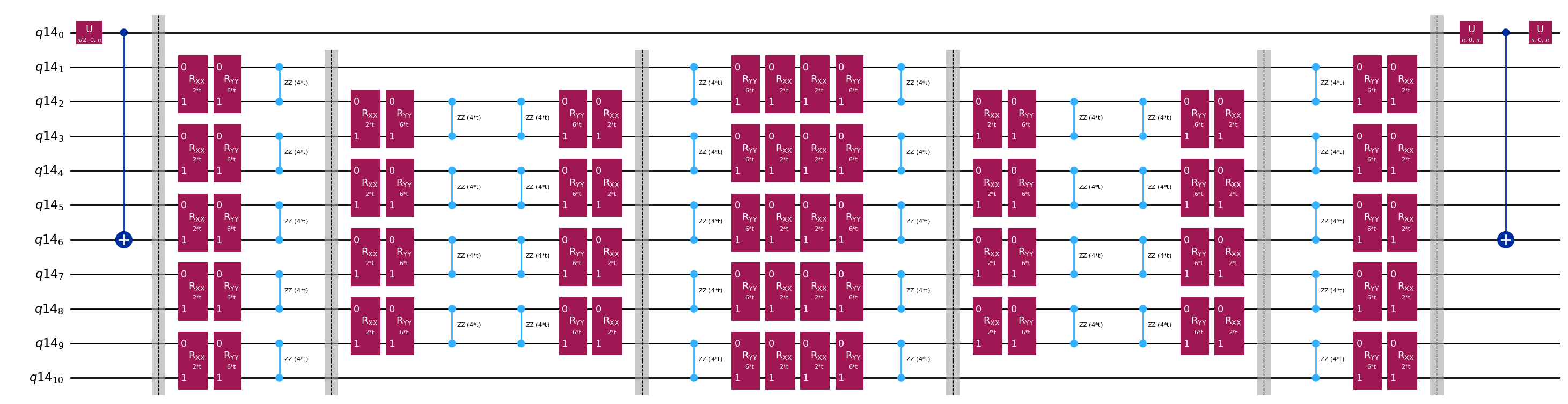
print(
"The optimized circuit has 2Q gates depth: ",
qc.decompose().decompose().depth(lambda x: x[0].num_qubits == 2),
)
The optimized circuit has 2Q gates depth: 50
Esta profundidad es considerablemente menor en comparación con el test de Hadamard original. Esta profundidad es manejable en los ordenadores cuánticos modernos, aunque sigue siendo bastante alta. Necesitaremos usar mitigación de errores de última generación para obtener resultados útiles.
Selecciona un backend en el que ejecutar nuestro cálculo de Krylov cuántico, para poder transpilar nuestro circuito y ejecutarlo en ese ordenador cuántico.
# Use the least-busy backend or specify a quantum computer using the syntax commented out below.
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
# Or you may choose a specify backend and channel if necessary for your workflow.
# service = QiskitRuntimeService(channel="ibm_quantum_platform")
# backend = service.backend("ibm_fez")
Ahora transpilamos nuestros circuitos y operadores.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
basis_gates = list(target.operation_names)
pm = generate_preset_pass_manager(
optimization_level=3, backend=backend, basis_gates=basis_gates
)
qc_trans = pm.run(qc)
print(qc_trans.depth(lambda x: x[0].num_qubits == 2))
print(qc_trans.count_ops())
qc_trans.draw("mpl", fold=-1, idle_wires=False, scale=0.5)
36
OrderedDict([('rz', 410), ('sx', 361), ('cz', 156), ('x', 18), ('barrier', 6)])

Después de la optimización, la profundidad de puertas de 2 qubits del circuito transpilado se reduce aún más.
4.3 Paso 3. Ejecutar con una primitiva de Qiskit Runtime
Ahora creamos PUBs para su ejecución con Estimator.
# Define observables to measure for S
observable_S_real = "I" * (n_qubits) + "X"
observable_S_imag = "I" * (n_qubits) + "Y"
observable_op_real = SparsePauliOp(
observable_S_real
) # define a sparse pauli operator for the observable
observable_op_imag = SparsePauliOp(observable_S_imag)
layout = qc_trans.layout # get layout of transpiled circuit
observable_op_real = observable_op_real.apply_layout(
layout
) # apply physical layout to the observable
observable_op_imag = observable_op_imag.apply_layout(layout)
observable_S_real = (
observable_op_real.paulis.to_labels()
) # get the label of the physical observable
observable_S_imag = observable_op_imag.paulis.to_labels()
observables_S = [[observable_S_real], [observable_S_imag]]
# Define observables to measure for H
# Hamiltonian terms to measure
observable_list = []
for pauli, coeff in zip(H_op.paulis, H_op.coeffs):
# print(pauli)
observable_H_real = pauli[::-1].to_label() + "X"
observable_H_imag = pauli[::-1].to_label() + "Y"
observable_list.append([observable_H_real])
observable_list.append([observable_H_imag])
layout = qc_trans.layout
observable_trans_list = []
for observable in observable_list:
observable_op = SparsePauliOp(observable)
observable_op = observable_op.apply_layout(layout)
observable_trans_list.append([observable_op.paulis.to_labels()])
observables_H = observable_trans_list
# Define a sweep over parameter values
params = np.vstack(parameters).T
# Estimate the expectation value for all combinations of
# observables and parameter values, where the pub result will have
# shape (# observables, # parameter values).
pub = (qc_trans, observables_S + observables_H, params)
Los circuitos para son calculables clásicamente. Llevamos esto a cabo antes de pasar al caso usando un ordenador cuántico.
from qiskit.quantum_info import StabilizerState, Pauli
qc_cliff = qc.assign_parameters({t: 0})
# Get expectation values from experiment
S_expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "X")
)
S_expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "Y")
)
# Get expectation values
S_expval = S_expval_real + 1j * S_expval_imag
H_expval = 0
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Get expectation values from experiment
expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "X")
)
expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "Y")
)
expval = expval_real + 1j * expval_imag
# Fill-in matrix elements
H_expval += coeff * expval
print(H_expval)
(10+0j)
Aunque logramos reducir la profundidad de puertas en varios órdenes de magnitud gracias al test de Hadamard eficiente, la profundidad sigue siendo suficiente como para requerir mitigación de errores de última generación. A continuación, especificamos los atributos de la mitigación utilizada. Todos los métodos empleados son importantes, pero vale la pena destacar específicamente la amplificación probabilística de errores (PEA). Esta poderosa técnica conlleva una gran sobrecarga cuántica. El cálculo realizado aquí puede tardar 20 minutos o más en ejecutarse en un ordenador cuántico real. Puede que quieras ajustar los parámetros a continuación para aumentar o disminuir la precisión y, en consecuencia, la sobrecarga. Los ajustes predeterminados a continuación producen resultados de alta fidelidad.
# Experiment options
num_randomizations = 300
num_randomizations_learning = 20
max_batch_circuits = 20
shots_per_randomization = 100
learning_pair_depths = [0, 4, 24]
noise_factors = [1, 1.3, 1.6]
# Base option formatting
options = {
# Builtin resilience settings for ZNE
"resilience": {
"measure_mitigation": True,
"zne_mitigation": True,
"zne": {"noise_factors": noise_factors},
# TREX noise learning configuration
"measure_noise_learning": {
"num_randomizations": num_randomizations_learning,
"shots_per_randomization": shots_per_randomization,
},
# PEA noise model configuration
"layer_noise_learning": {
"max_layers_to_learn": 10,
"layer_pair_depths": learning_pair_depths,
"shots_per_randomization": shots_per_randomization,
"num_randomizations": num_randomizations_learning,
},
},
# Randomization configuration
"twirling": {
"num_randomizations": num_randomizations,
"shots_per_randomization": shots_per_randomization,
"strategy": "all",
},
# Experimental settings for PEA method
"experimental": {
# # Just in case, disable any further qiskit transpilation not related to twirling / DD
# "skip_transpilation": True,
# Execution configuration
"execution": {
"max_pubs_per_batch_job": max_batch_circuits,
"fast_parametric_update": True,
},
# Error Mitigation configuration
"resilience": {
# ZNE Configuration
"zne": {
"amplifier": "pea",
"return_all_extrapolated": True,
"return_unextrapolated": True,
"extrapolated_noise_factors": [0] + noise_factors,
}
},
},
}
Finalmente, ejecutamos los circuitos para y con Estimator.
# This job required 17 minutes of QPU time to run on a Heron r2 processor. This is only an estimate.
# Your execution time may vary.
with Batch(backend=backend) as batch:
# Estimator
estimator = Estimator(mode=batch, options=options)
job = estimator.run([pub], precision=1)
4.4 Paso 4. Posprocesar y analizar resultados
Lo que hemos obtenido del ordenador cuántico son los elementos de matriz individuales de y los grupos de Pauli que conmutan y que forman los elementos de matriz de . Estos términos deben combinarse para recuperar nuestras matrices y así poder resolver el problema de valores propios generalizado.
# Store the outputs as 'results'.
results = job.result()[0]
Calcular las matrices del Hamiltoniano efectivo y de solapamiento
Primero calculamos la fase acumulada por el estado durante la evolución temporal no controlada.
prefactors = [
np.exp(-1j * sum([c for p, c in H_op.to_list() if "Z" in p]) * i * dt)
for i in range(1, krylov_dim)
]
Una vez que tenemos los resultados de las ejecuciones de los circuitos, podemos posprocesar los datos para calcular los elementos de matriz de .
# Assemble S, the overlap matrix of dimension D:
S_first_row = np.zeros(krylov_dim, dtype=complex)
S_first_row[0] = 1 + 0j
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[0][0][i] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[1][0][i] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
S_first_row[i + 1] += prefactors[i] * expval
S_first_row_list = S_first_row.tolist() # for saving purposes
S_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in it.product(range(krylov_dim), repeat=2):
if i >= j:
S_circ[j, i] = S_first_row[i - j]
else:
S_circ[j, i] = np.conj(S_first_row[j - i])
from sympy import Matrix
Matrix(S_circ)
Y los elementos de matriz de
import itertools
# Assemble S, the overlap matrix of dimension D:
H_first_row = np.zeros(krylov_dim, dtype=complex)
H_first_row[0] = H_expval
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[2 + 2 * obs_idx][0][
i
] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[2 + 2 * obs_idx + 1][0][
i
] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
H_first_row[i + 1] += prefactors[i] * coeff * expval
H_first_row_list = H_first_row.tolist()
H_eff_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in itertools.product(range(krylov_dim), repeat=2):
if i >= j:
H_eff_circ[j, i] = H_first_row[i - j]
else:
H_eff_circ[j, i] = np.conj(H_first_row[j - i])
from sympy import Matrix
Matrix(H_eff_circ)
Finalmente, podemos resolver el problema de valores propios generalizado para :
y obtener una estimación de la energía del estado fundamental
gnd_en_circ_est_list = []
for d in range(1, krylov_dim + 1):
# Solve generalized eigenvalue problem
gnd_en_circ_est = solve_regularized_gen_eig(
H_eff_circ[:d, :d], S_circ[:d, :d], threshold=1e-1
)
gnd_en_circ_est_list.append(gnd_en_circ_est)
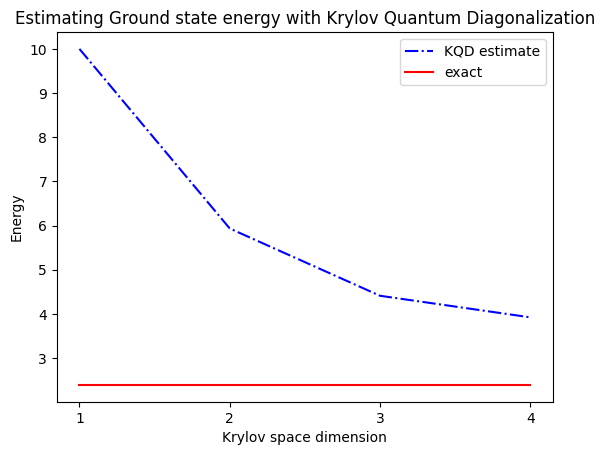
print("The estimated ground state energy is: ", gnd_en_circ_est)
The estimated ground state energy is: 10.0
The estimated ground state energy is: 5.933953916292923
The estimated ground state energy is: 4.4101773995740645
The estimated ground state energy is: 3.921288588521255
Para un sector de una sola partícula, podemos calcular eficientemente el estado fundamental de este sector del Hamiltoniano de forma clásica.
gs_en = single_particle_gs(H_op, n_qubits)
n_sys_qubits 10
n_exc 1 , subspace dimension 11
single particle ground state energy: 2.391547869638771
len(H_op)
27
plt.plot(
range(1, krylov_dim + 1),
gnd_en_circ_est_list,
color="blue",
linestyle="-.",
label="KQD estimate",
)
plt.plot(
range(1, krylov_dim + 1),
[gs_en] * krylov_dim,
color="red",
linestyle="-",
label="exact",
)
plt.xticks(range(1, krylov_dim + 1), range(1, krylov_dim + 1))
plt.legend()
plt.xlabel("Krylov space dimension")
plt.ylabel("Energy")
plt.title("Estimating Ground state energy with Krylov Quantum Diagonalization")
plt.show()
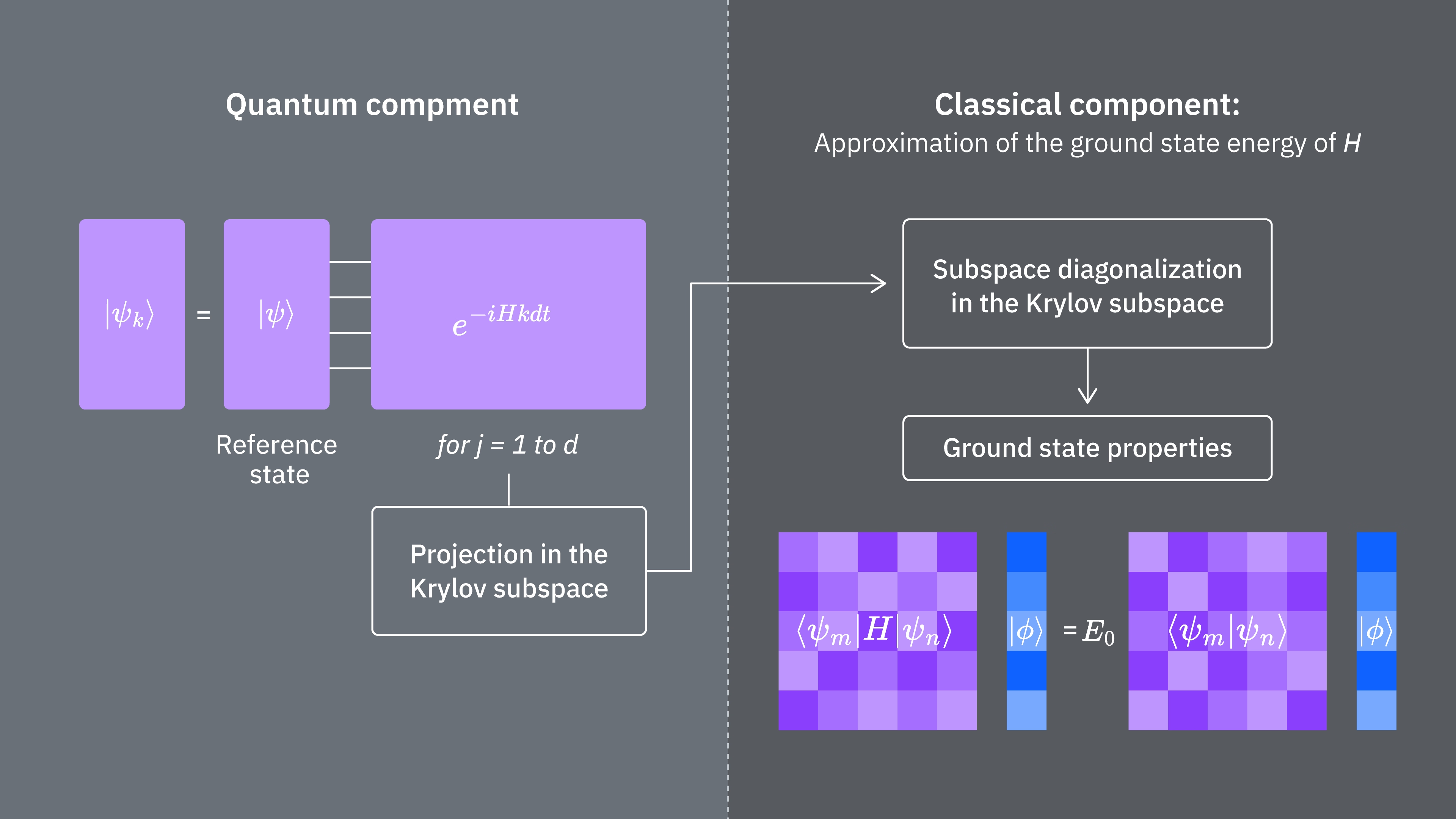
5. Discusión y extensión
Para resumir, comenzamos con un estado de referencia, luego lo evolucionamos durante diferentes periodos de tiempo para generar el subespacio de Krylov unitario. Proyectamos nuestro Hamiltoniano en ese subespacio. También estimamos los solapamientos de los vectores del subespacio. Finalmente, resolvemos clásicamente el problema de valores propios generalizado de menor dimensión.
Comparemos qué determina los costos computacionales de usar la técnica de Krylov de forma clásica y cuántica. No existen analogías perfectas entre los enfoques clásico y cuántico para todos los pasos. Esta tabla recopila algunos escalados de los distintos pasos para su consideración.
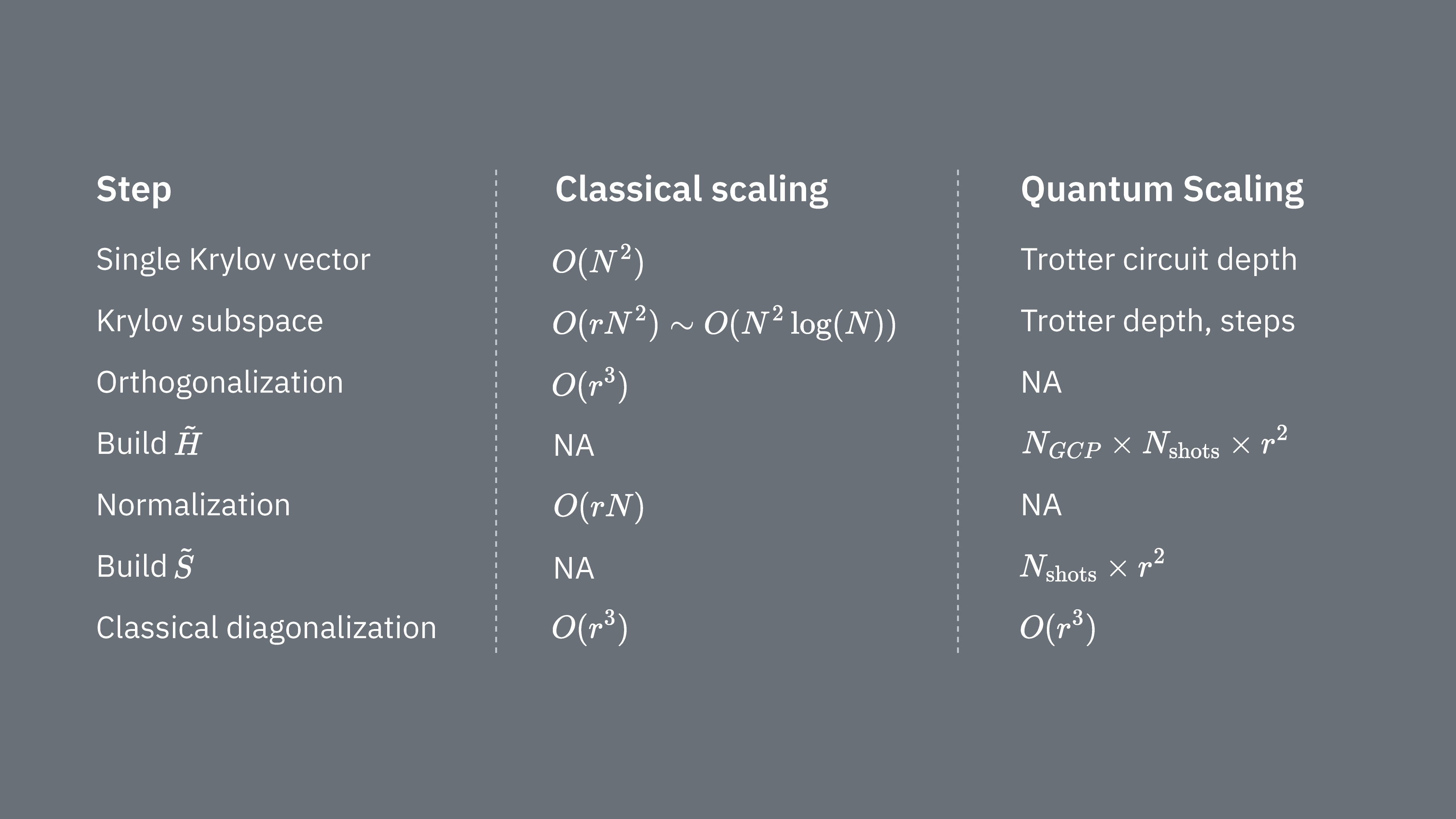
Recuerda que los Hamiltonianos generalmente tienen términos que no se pueden medir simultáneamente (porque no conmutan entre sí). Agrupamos los términos del Hamiltoniano en grupos de operadores de Pauli conmutantes que pueden medirse todos a la vez, y es posible que necesitemos muchos de estos grupos para dar cuenta de todos los términos que no conmutan entre sí. Para construir en un computador cuántico se requieren mediciones separadas para cada grupo de cadenas de Pauli conmutantes del Hamiltoniano, y cada una de ellas requiere muchas mediciones (shots). Debemos hacer esto para elementos de la matriz distintos, correspondientes a combinaciones de diferentes factores de evolución temporal. A veces hay formas de reducir esto, pero en este tratamiento aproximado, el tiempo requerido para esto escala como Los elementos de deben estimarse, lo que escala como . Finalmente, resolver el problema de valores propios generalizado en el espacio proyectado, clásicamente, toma
Vemos que la diagonalización cuántica de Krylov puede ser útil en casos donde el número de grupos de Pauli conmutantes en el Hamiltoniano es relativamente pequeño. Estas dependencias de escalado sugieren algunas aplicaciones donde el método de Krylov puede ser útil, y otras donde probablemente no lo será. Algunos Hamiltonianos tienen alta complejidad cuando se mapean a qubits, involucrando muchas cadenas de Pauli no conmutantes que no pueden particionarse fácilmente en unos pocos grupos conmutantes. Esto ocurre frecuentemente en problemas de química cuántica, por ejemplo. Esta complejidad presenta dos desafíos principales para los computadores cuánticos de corto plazo:
- La estimación de cada elemento de se vuelve costosa computacionalmente debido al gran número de términos.
- Los circuitos de Trotter requeridos se vuelven prohibitivamente profundos.
Ambos puntos anteriores serán menos problemáticos cuando los computadores cuánticos alcancen la tolerancia a fallos, pero deben tenerse en cuenta a corto plazo. Incluso los sistemas con mapeos "más simples" que los de la química cuántica pueden experimentar los mismos impedimentos, si los Hamiltonianos tienen demasiados términos no conmutantes. El método de Krylov es más útil cuando el Hamiltoniano puede particionarse en relativamente pocos grupos de Pauli conmutantes, y cuando es fácil de implementar en circuitos de Trotter. Ambas condiciones se satisfacen, por ejemplo, en muchos modelos de red de interés en física. KQD es especialmente útil cuando se sabe muy poco sobre el estado fundamental. Esto se debe a sus garantías de convergencia inherentes y a su aplicabilidad en escenarios donde los métodos alternativos son inviables por falta de conocimiento del estado fundamental.
Aunque KQD es una herramienta poderosa, los aspectos más lentos del protocolo — en particular la estimación de cada elemento del Hamiltoniano proyectado y el solapamiento de los estados de Krylov — representan oportunidades de mejora. Un enfoque alternativo consiste en combinar los métodos de Krylov con métodos basados en muestreo, que son el tema de la siguiente lección.
6. Apéndices
Apéndice I: Subespacio de Krylov a partir de evoluciones temporales reales
El espacio de Krylov unitario se define como
para algún paso de tiempo que determinaremos más adelante. Supón temporalmente que es par: entonces define . Observa que cuando proyectamos el Hamiltoniano en el subespacio de Krylov anterior, es indistinguible del subespacio de Krylov
es decir, donde todas las evoluciones temporales están desplazadas pasos hacia atrás. La razón por la que es indistinguible es que los elementos de la matriz
son invariantes bajo desplazamientos globales del tiempo de evolución, ya que las evoluciones temporales conmutan con el Hamiltoniano. Para impar, podemos usar el análisis para .
Queremos demostrar que en algún lugar de este subespacio de Krylov está garantizado que existe un estado de baja energía. Lo hacemos mediante el siguiente resultado, que se deriva del Teorema 3.1 en [3]:
Afirmación 1: existe una función tal que para energías en el rango espectral del Hamiltoniano (es decir, entre la energía del estado fundamental y la energía máxima)...
- para todos los valores de que están alejados de , es decir, está suprimida exponencialmente
- es una combinación lineal de para
A continuación damos una demostración, pero puede omitirse sin problema a menos que se quiera entender el argumento completo y riguroso. Por ahora nos centramos en las implicaciones de la afirmación anterior. Por la propiedad 3 anterior, podemos ver que el subespacio de Krylov desplazado contiene el estado . Este es nuestro estado de baja energía. Para ver por qué, escribamos en la base de autoenergías:
donde es el k-ésimo autoestado de energía y es su amplitud en el estado inicial . Expresado en estos términos, viene dado por
usando el hecho de que podemos reemplazar por cuando actúa sobre el autoestado . El error de energía de este estado es, por tanto,
Para convertir esto en una cota superior más fácil de entender, primero separamos la suma del numerador en términos con y términos con :
Podemos acotar superiormente el primer término por ,
donde el primer paso se sigue porque para cada en la suma, y el segundo paso se sigue porque la suma del numerador es un subconjunto de la suma del denominador. Para el segundo término, primero acotamos inferiormente el denominador por , ya que : juntando todo de nuevo, esto da
Para simplificar lo que queda, observa que para todos estos , por la definición de sabemos que . Acotando adicionalmente y acotando se obtiene
Esto se cumple para cualquier , de modo que si establecemos igual al error objetivo, entonces la cota de error anterior converge hacia ese valor exponencialmente con la dimensión de Krylov . Observa también que si , entonces el término desaparece completamente en la cota anterior.
Para completar el argumento, primero observamos que lo anterior es solo el error de energía del estado particular , en lugar del error de energía del estado de menor energía en el subespacio de Krylov. Sin embargo, por el principio variacional (de Rayleigh-Ritz), el error de energía del estado de menor energía en el subespacio de Krylov está acotado superiormente por el error de energía de cualquier estado en el subespacio de Krylov, por lo que lo anterior también es una cota superior sobre el error de energía del estado de menor energía, es decir, la salida del algoritmo de diagonalización cuántica de Krylov.
Un análisis similar al anterior puede llevarse a cabo que además tenga en cuenta el ruido y el procedimiento de umbralización discutido en el notebook. Consulta [2] y [4] para ese análisis.
Apéndice II: demostración de la Afirmación 1
Lo siguiente se deriva principalmente de [3], Teorema 3.1: sean y sea el espacio de polinomios residuales (polinomios cuyo valor en 0 es 1) de grado como máximo . La solución a
es
y el valor mínimo correspondiente es
Queremos convertir esto en una función que pueda expresarse naturalmente en términos de exponenciales complejas, porque esas son las evoluciones temporales reales que generan el subespacio cuántico de Krylov. Para ello, es conveniente introducir la siguiente transformación de energías dentro del rango espectral del Hamiltoniano a números en el rango : define
donde es un paso de tiempo tal que . Observa que y crece a medida que se aleja de .
Ahora usando el polinomio con los parámetros a, b, d establecidos a , , y d = int(r/2), definimos la función:
donde es la energía del estado fundamental. Podemos ver insertando que es un polinomio trigonométrico de grado , es decir, una combinación lineal de para . Además, a partir de la definición de anterior tenemos que y para cualquier en el rango espectral tal que tenemos
Referencias:
[1] https://arxiv.org/abs/2407.14431
[2] https://arxiv.org/abs/1811.09025
[3] https://people.math.ethz.ch/~mhg/pub/biksm.pdf
[4] https://academic.oup.com/book/36426
[5] https://en.wikipedia.org/wiki/Krylov_subspace
[6] Krylov Subspace Methods: Principles and Analysis, Jörg Liesen, Zdenek Strakos https://academic.oup.com/book/36426
[7] Iterative Methods for Sparse Linear Systems" by Yousef Saad
[8] "MINRES-QLP: A Krylov Subspace Method for Indefinite or Singular Symmetric Systems" by Sou-Cheng Choi, Christopher Paige, and Michael Saunders (https://epubs.siam.org/doi/10.1137/100787921)
[9] Ethan N. Epperly, Lin Lin, and Yuji Nakatsukasa. "A theory of quantum subspace diagonalization". SIAM Journal on Matrix Analysis and Applications 43, 1263–1290 (2022).