Diagonalización cuántica basada en muestras de un hamiltoniano de química
Estimación de uso: menos de un minuto en un procesador Heron r2 (NOTA: Esto es solo una estimación. El tiempo de ejecución real puede variar.)
Resultados de aprendizaje
Después de completar este tutorial, los usuarios deberían entender:
- Cómo usar el complemento SQD de Qiskit para aproximar la energía del estado fundamental de un sistema molecular usando cadenas de bits muestreadas desde una unidad de procesamiento cuántico (QPU).
- Cómo usar ffsim para construir un circuito de ansatz de Jastrow con clúster unitario local (LUCJ) para simulación de química cuántica.
Prerrequisitos
Recomendamos que los usuarios se familiaricen con los siguientes temas antes de realizar este tutorial:
- Química cuántica y segunda cuantización
- Uso de la primitiva Sampler para muestrear circuitos cuánticos
Antecedentes
En este tutorial, mostramos cómo post-procesar muestras cuánticas ruidosas para aproximar el estado fundamental de la molécula de nitrógeno en la longitud de enlace de equilibrio, usando el complemento SQD de Qiskit para implementar el algoritmo de diagonalización cuántica basada en muestras (SQD). Más detalles sobre el software se pueden encontrar en la documentación correspondiente, incluyendo un ejemplo sencillo para comenzar.
Este tutorial es recomendado para usuarios familiarizados con química cuántica: específicamente, familiaridad con encontrar energías del estado fundamental de una molécula. Para un recorrido detallado del flujo de trabajo, consulta el curso de algoritmos de diagonalización cuántica.
SQD es una técnica para encontrar valores propios y vectores propios de operadores cuánticos, como un hamiltoniano de un sistema cuántico, usando computación cuántica y computación clásica distribuida de manera conjunta. La computación clásica distribuida se usa para procesar muestras obtenidas de un procesador cuántico, y para proyectar y diagonalizar un hamiltoniano objetivo en un subespacio que éstas abarcan. Un flujo de trabajo basado en SQD tiene los siguientes pasos:
- Elegir un ansatz de circuito y aplicarlo en una computadora cuántica a un estado de referencia (en este caso, el estado de Hartree-Fock).
- Muestrear cadenas de bits del estado cuántico resultante.
- Ejecutar el procedimiento de recuperación autoconsistente de configuraciones en las cadenas de bits para obtener la aproximación al estado fundamental.
Se sabe que SQD funciona bien cuando el estado propio objetivo es disperso: la función de onda está soportada en un conjunto de estados base cuyo tamaño no crece exponencialmente con el tamaño del problema.
Química cuántica
El hamiltoniano de un sistema molecular puede escribirse como
donde los y son números complejos llamados integrales moleculares que pueden calcularse a partir de la especificación de la molécula usando un programa de computadora. En este tutorial, calculamos las integrales usando el paquete de software PySCF.
Para detalles sobre cómo se deriva el hamiltoniano molecular, consulta un libro de texto de química cuántica (por ejemplo, Modern Quantum Chemistry de Szabo y Ostlund). Para una explicación de alto nivel sobre cómo los problemas de química cuántica se mapean en computadoras cuánticas, consulta la conferencia Mapping Problems to Qubits de la Qiskit Global Summer School 2024.
Ansatz de Jastrow con clúster unitario local (LUCJ)
SQD requiere un ansatz de circuito cuántico del cual tomar muestras. En este tutorial, usaremos el ansatz de Jastrow con clúster unitario local (LUCJ) debido a su combinación de motivación física y compatibilidad con el hardware. Usaremos ffsim para construir el circuito del ansatz.
El ansatz LUCJ se adapta a las QPUs con conectividad de qubits restringida. Los orbitales de espín se mapean a qubits de modo que el ansatz no requiera enrutamiento con puertas SWAP. El hardware de IBM® tiene una topología de qubits en rejilla hexagonal pesada (heavy-hex), en cuyo caso podemos adoptar un patrón de "zigzag", representado a continuación. En este patrón, los orbitales con el mismo espín se mapean a qubits con una topología lineal (círculos rojos y azules), y una conexión entre orbitales de diferente espín está presente cada 4 orbitales espaciales, siendo la conexión facilitada por un qubit auxiliar (círculos morados).
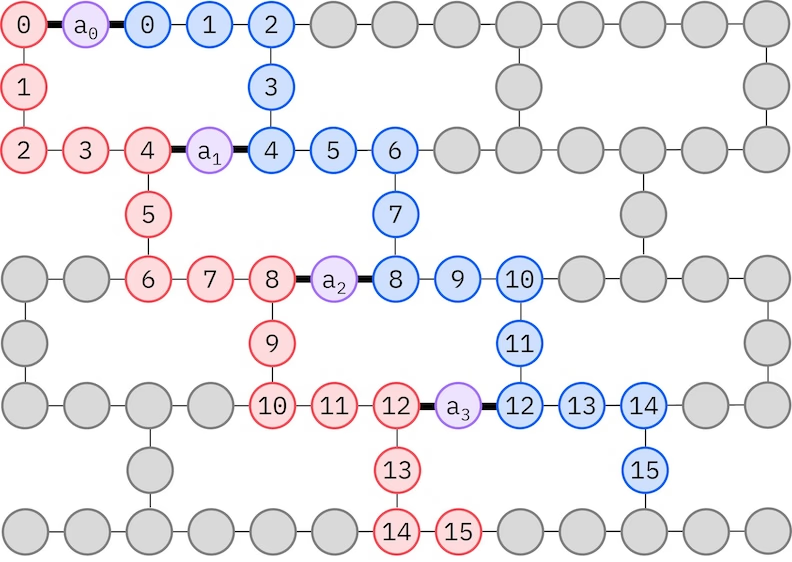
Recuperación autoconsistente de configuraciones
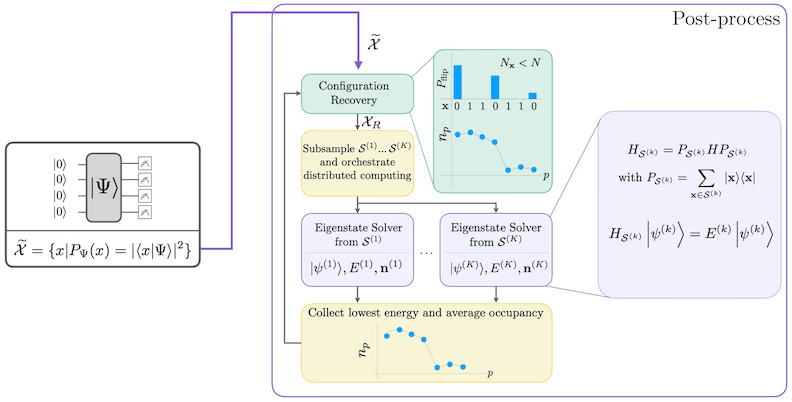
El procedimiento de recuperación autoconsistente de configuraciones está diseñado para extraer la mayor cantidad de señal posible de las muestras cuánticas ruidosas. Dado que el hamiltoniano molecular conserva el número de partículas y el espín Z, tiene sentido elegir un ansatz de circuito que también conserve estas simetrías. Cuando se aplica al estado de Hartree-Fock, el estado resultante tiene un número de partículas y espín Z fijos en el caso sin ruido. Por lo tanto, las mitades de espín- y espín- de cualquier cadena de bits muestreada de este estado deberían tener el mismo peso de Hamming que en el estado de Hartree-Fock. Debido a la presencia de ruido en los procesadores cuánticos actuales, algunas cadenas de bits medidas violarán esta propiedad. Una forma simple de postselección descartaría estas cadenas de bits, pero esto es un desperdicio porque esas cadenas de bits aún podrían contener alguna señal. El procedimiento de recuperación autoconsistente intenta recuperar parte de esa señal en el post-procesamiento. El procedimiento es iterativo y requiere como entrada una estimación de las ocupaciones promedio de cada orbital en el estado fundamental, que primero se calcula a partir de las muestras crudas. El procedimiento se ejecuta en un bucle, y cada iteración tiene los siguientes pasos:
- Para cada cadena de bits que viola las simetrías especificadas, invertir sus bits con un procedimiento probabilístico diseñado para acercar la cadena de bits a la estimación actual de las ocupaciones orbitales promedio, para obtener una nueva cadena de bits.
- Recopilar todas las cadenas de bits antiguas y nuevas que satisfagan las simetrías, y submuestrear subconjuntos de un tamaño fijo, elegido de antemano.
- Para cada subconjunto de cadenas de bits, proyectar el hamiltoniano en el subespacio generado por los vectores base correspondientes (consulta la sección anterior para una descripción de estos vectores base), y calcular una estimación del estado fundamental del hamiltoniano proyectado en una computadora clásica.
- Actualizar la estimación de las ocupaciones orbitales promedio con la estimación del estado fundamental que tenga la energía más baja.
Diagrama del flujo de trabajo de SQD
El flujo de trabajo de SQD se representa en el siguiente diagrama:
Requisitos
Antes de comenzar este tutorial, asegúrate de tener instalado lo siguiente:
- Qiskit SDK v1.0 o posterior, con soporte de visualización
- Qiskit Runtime v0.22 o posterior (
pip install qiskit-ibm-runtime) - Complemento SQD de Qiskit v0.11 o posterior (
pip install qiskit-addon-sqd) - ffsim v0.0.75 o posterior (
pip install ffsim)
Configuración
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Ejemplo de simulador a pequeña escala
En este tutorial, encontraremos una aproximación al estado fundamental de una molécula de nitrógeno cerca de su distancia de enlace de equilibrio. Primero usamos un conjunto de bases STO-6G pequeño para poder simular el experimento y verificar que funciona.
Paso 1: Mapear las entradas clásicas a un problema cuántico
Primero, especificamos la molécula y sus propiedades.
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
Antes de construir el circuito del ansatz LUCJ, primero realizamos un cálculo CCSD en la siguiente celda de código. Las amplitudes y de este cálculo se usarán para inicializar los parámetros del ansatz.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
Ahora, usamos ffsim para crear el circuito del ansatz. Dado que nuestra molécula tiene un estado de Hartree-Fock de capa cerrada, usamos la variante con equilibrio de espín del ansatz UCJ, UCJOpSpinBalanced. Establecemos optimize=True en el método from_t_amplitudes para habilitar la doble factorización "comprimida" de las amplitudes (consulta The local unitary cluster Jastrow (LUCJ) ansatz en la documentación de ffsim para más detalles).
Dado que el ansatz LUCJ se adapta a la conectividad disponible de la QPU, tenemos que inicializar el backend de la QPU antes de crear el ansatz. Por ahora, crearemos un backend genérico con un mapa de acoplamiento heavy-hex y un conjunto de puertas al que el ansatz LUCJ se descompone naturalmente. Luego, usaremos ffsim.qiskit.generate_lucj_pass_manager para crear un gestor de pasadas especializado en transpilar el ansatz LUCJ al backend dado según el diseño de "zigzag" descrito en la sección de antecedentes sobre el ansatz LUCJ. Esta función usa una heurística de puntuación para minimizar los errores asociados con el diseño seleccionado, lo cual es importante si tu backend es una QPU real, o un simulador con un modelo de ruido. Además de devolver el gestor de pasadas, esta función también devuelve los pares de acoplamiento alfa-beta que pueden implementarse en el hardware. Si no se pueden implementar todos los pares, emite una advertencia.
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Paso 2: Optimizar para la ejecución en hardware cuántico
A continuación, optimizamos el circuito para un hardware objetivo. Normalmente, este paso implica inicializar el backend de hardware y un gestor de pasadas para ese backend. Sin embargo, dado que el ansatz LUCJ se adapta a la conectividad del hardware, ya realizamos estas acciones en el paso anterior. Solo queda ejecutar el gestor de pasadas en el circuito para transpilarlo a un circuito ISA que pueda ejecutarse directamente en la QPU.
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
Paso 3: Ejecutar usando primitivas de Qiskit
Después de optimizar el circuito para la ejecución en hardware, estamos listos para ejecutarlo en el hardware objetivo y recopilar muestras para la estimación de la energía del estado fundamental. Como solo tenemos un circuito, usaremos el modo de ejecución de trabajos de Qiskit Runtime y ejecutaremos nuestro circuito.
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
Paso 4: Post-procesar y devolver el resultado en el formato clásico deseado
Una métrica útil para juzgar la calidad de la salida de la QPU es el número de configuraciones válidas devueltas. Una configuración válida tiene el número de partículas y espín Z correctos, lo que significa que la mitad derecha de la cadena de bits tiene peso de Hamming igual al número de electrones con espín hacia arriba, y la mitad izquierda tiene peso de Hamming igual al número de electrones con espín hacia abajo. La siguiente celda calcula la fracción de configuraciones muestreadas que son válidas.
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
Todas las cadenas de bits son válidas porque estamos muestreando el circuito en un simulador sin ruido. Al ejecutar en una QPU ruidosa, la fracción será menor que uno, pero esperamos que sea mayor que la fracción que esperarías si las cadenas de bits se muestrearan uniformemente al azar, lo cual se calcula en la siguiente celda.
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
Ahora, estimamos la energía del estado fundamental del hamiltoniano usando la función diagonalize_fermionic_hamiltonian. Esta función realiza el procedimiento de recuperación autoconsistente de configuraciones para refinar iterativamente las muestras cuánticas ruidosas y mejorar la estimación de energía. Pasamos una función de callback para poder guardar los resultados intermedios para su análisis posterior. Consulta la documentación de la API para obtener explicaciones de los argumentos de diagonalize_fermionic_hamiltonian.
Aquí, usamos el argumento initial_occupancies de diagonalize_fermionic_hamiltonian para especificar la configuración de Hartree-Fock como la suposición inicial para las ocupaciones orbitales en el estado fundamental. Este enfoque es razonable para sistemas donde el estado fundamental tiene un soporte significativo en la configuración de Hartree-Fock, pero puede no ser apropiado en otras situaciones, aunque métodos computacionales más avanzados podrían ofrecer mejores suposiciones iniciales en esos casos. Especificar initial_occupancies también permite que la recuperación de configuraciones funcione incluso si no se muestrearon configuraciones válidas, como puede ocurrir al muestrear un circuito grande en una QPU ruidosa. Sin este argumento, la recuperación de configuraciones fallaría y generaría un error si no se proporcionan configuraciones válidas.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
Visualizar los resultados
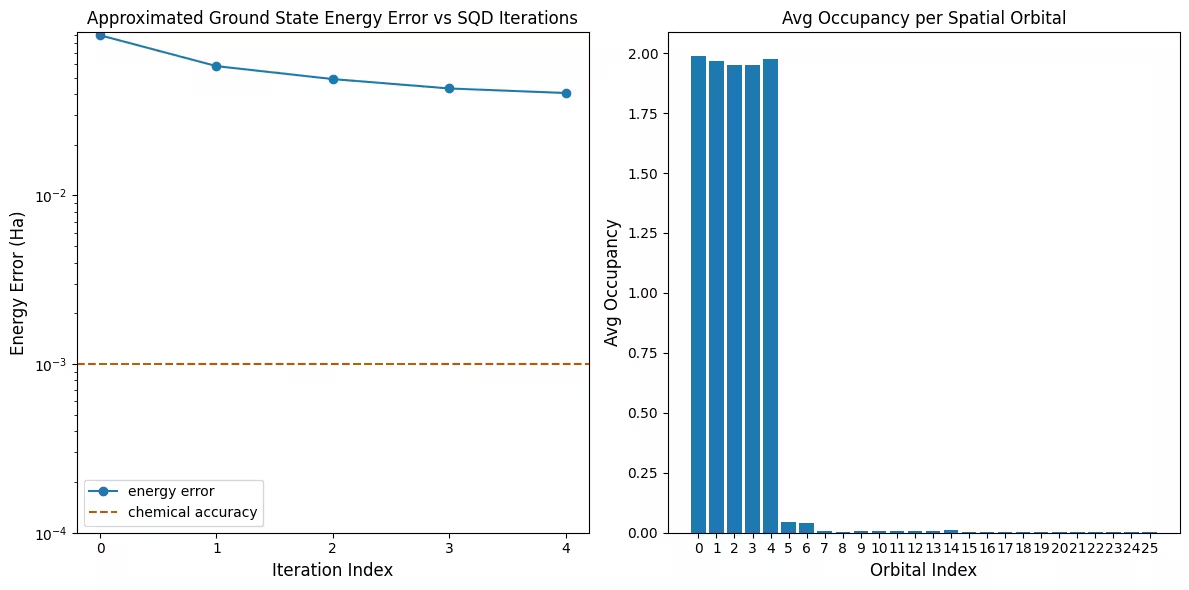
El primer gráfico muestra que en esta simulación ya estamos dentro de 1 mH de la respuesta exacta después de la primera iteración (la precisión química se acepta típicamente como 1 kcal/mol 1.6 mH). Sin embargo, este es un sistema pequeño y, dado que las muestras no tienen ruido, no se necesita recuperación de configuraciones. En un sistema más grande ejecutado en una QPU ruidosa, podrían necesitarse múltiples iteraciones de recuperación de configuraciones, y la precisión final podría ser peor. En general, la energía puede mejorarse permitiendo más iteraciones de recuperación de configuraciones o aumentando el número de muestras por lote.
El segundo gráfico muestra la ocupación promedio de cada orbital espacial después de la iteración final. Podemos ver que tanto los electrones con espín hacia arriba como con espín hacia abajo ocupan los primeros cinco orbitales con alta probabilidad en nuestras soluciones.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Ejemplo de hardware a gran escala
Ahora, ejecutamos un ejemplo más grande en hardware cuántico real. Aquí, derivaremos un espacio activo para la molécula de nitrógeno del conjunto de bases cc-pVDZ.
Pasos 1-4
Aquí reunimos todos los pasos en un único flujo de trabajo a mayor escala, que luego se ejecuta en hardware cuántico real.
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
Siguientes pasos
Si este trabajo te pareció interesante, podrías estar interesado en el siguiente material:
- Diagonalización cuántica de Krylov basada en muestras de un modelo de red fermiónica - un tutorial relacionado que usa circuitos de evolución temporal en lugar de un ansatz variacional
- Escalar flujos de trabajo de química SQD con el solver Dice - una página que muestra cómo usar el software Dice más eficiente para la diagonalización
- Documentación de la API del complemento SQD - referencia de la función
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer - el artículo en el que se basa este tutorial