Diagonalización cuántica de Krylov basada en muestras de un modelo de red fermiónica
Estimación de uso: Nueve segundos en un procesador Heron r2 (NOTA: Esta es solo una estimación. Tu tiempo de ejecución puede variar.)
Resultados de aprendizaje
Después de completar este tutorial, los usuarios deberían comprender:
- Cómo usar el complemento SQD de Qiskit para aproximar la energía del estado fundamental de un modelo de red usando cadenas de bits muestreadas de una unidad de procesamiento cuántico (QPU).
- Cómo usar ffsim para construir circuitos de evolución temporal para simulación fermiónica.
- Cómo combinar muestras de múltiples circuitos para el posprocesamiento con el algoritmo de diagonalización cuántica de Krylov basada en muestras (SQKD).
Prerequisitos
Sugerimos que los usuarios estén familiarizados con los siguientes temas antes de completar este tutorial:
- Diagonalización cuántica basada en muestras de un hamiltoniano químico
- Diagonalización cuántica de Krylov de hamiltonianos de red
- Primitivas de Qiskit
Antecedentes
Este tutorial muestra cómo usar la diagonalización cuántica basada en muestras (SQD) para estimar la energía del estado fundamental de un modelo de red fermiónica. Específicamente, estudiamos el modelo de Anderson de impureza única (SIAM) unidimensional, que se usa para describir impurezas magnéticas embebidas en metales.
Este tutorial sigue un flujo de trabajo similar al tutorial relacionado Diagonalización cuántica basada en muestras de un hamiltoniano químico. Sin embargo, una diferencia clave radica en cómo se construyen los circuitos cuánticos. El otro tutorial usa un ansatz variacional heurístico, que es atractivo para hamiltonianos químicos con potencialmente millones de términos de interacción. Por otro lado, este tutorial usa circuitos que aproximan la evolución temporal por el hamiltoniano. Dichos circuitos pueden ser profundos, lo que hace que este enfoque sea mejor para aplicaciones a modelos de red. Los vectores de estado preparados por estos circuitos forman la base de un subespacio de Krylov y, como resultado, el algoritmo converge de manera demostrable y eficiente al estado fundamental, bajo suposiciones adecuadas.
El enfoque utilizado en este tutorial puede verse como una combinación de las técnicas usadas en SQD y la diagonalización cuántica de Krylov (KQD). El enfoque combinado se denomina a veces diagonalización cuántica de Krylov basada en muestras (SQKD). Consulta Diagonalización cuántica de Krylov de hamiltonianos de red para ver un tutorial sobre el método KQD.
Este tutorial se basa en el trabajo "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization", al que puedes referirte para obtener más detalles.
Modelo de Anderson de impureza única (SIAM)
El hamiltoniano SIAM unidimensional es una suma de tres términos:
donde
Aquí, son los operadores de creación/aniquilación fermiónicos para el sitio de baño con espín , son los operadores de creación/aniquilación para el modo de impureza, y . , y son números reales que describen las interacciones de hopping, on-site e hibridación, y es un número real que especifica el potencial químico.
Observa que el hamiltoniano es una instancia específica del hamiltoniano genérico de electrones en interacción,
donde consiste en términos de un cuerpo, que son cuadráticos en los operadores de creación y aniquilación fermiónicos, y consiste en términos de dos cuerpos, que son cuárticos. Para el SIAM,
y contiene el resto de los términos en el hamiltoniano. Para representar el hamiltoniano de manera programática, almacenamos la matriz y el tensor .
Bases de posición y momento
Debido a la simetría traslacional aproximada en , no esperamos que el estado fundamental sea disperso en la base de posición (la base orbital en la que se especifica el hamiltoniano anteriormente). El rendimiento de SQD está garantizado solo si el estado fundamental es disperso, es decir, tiene peso significativo en solo un pequeño número de estados base computacionales. Para mejorar la dispersión del estado fundamental, realizamos la simulación en la base orbital en la que es diagonal. Llamamos a esta base la base de momento. Dado que es un hamiltoniano fermiónico cuadrático, puede diagonalizarse eficientemente mediante una rotación orbital.
Evolución temporal aproximada por el hamiltoniano
Para aproximar la evolución temporal por el hamiltoniano, usamos una descomposición de Trotter-Suzuki de segundo orden,
Bajo la transformación de Jordan-Wigner, la evolución temporal por equivale a una sola compuerta CPhase entre los orbitales de espín hacia arriba y hacia abajo en el sitio de impureza. Dado que es un hamiltoniano fermiónico cuadrático, la evolución temporal por equivale a una rotación orbital.
Los estados base de Krylov , donde es la dimensión del subespacio de Krylov, se forman mediante la aplicación repetida de un solo paso de Trotter, de modo que
En el siguiente flujo de trabajo basado en SQD, muestrearemos a partir de este conjunto de circuitos y procesaremos el conjunto combinado de cadenas de bits con SQD. Este enfoque contrasta con el utilizado en el tutorial relacionado Diagonalización cuántica basada en muestras de un hamiltoniano químico, donde las muestras se extrajeron de un único circuito variacional heurístico.
Requisitos
Antes de comenzar este tutorial, asegúrate de tener instalado lo siguiente:
- Qiskit SDK v1.0 o posterior, con soporte de visualización
- Qiskit Runtime v0.22 o posterior (
pip install qiskit-ibm-runtime) - SQD Qiskit addon v0.11 o posterior (
pip install qiskit-addon-sqd) - ffsim v0.0.72 o posterior (
pip install ffsim)
Ejemplo con simulador a pequeña escala
Paso 1: Mapear el problema a un circuito cuántico
Primero, generamos el hamiltoniano SIAM en la base de posición. El hamiltoniano se representa mediante la matriz y el tensor . Luego, lo rotamos a la base de momento. En la base de posición, colocamos la impureza en el primer sitio. Sin embargo, cuando rotamos a la base de momento, movemos la impureza a un sitio central para facilitar las interacciones con otros orbitales.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime scipy
import numpy as np
import pyscf.fci
def siam_hamiltonian(
norb: int,
hopping: float,
onsite: float,
hybridization: float,
chemical_potential: float,
) -> tuple[np.ndarray, np.ndarray]:
"""Hamiltonian for the single-impurity Anderson model."""
# Place the impurity on the first site
impurity_orb = 0
# One body matrix elements in the "position" basis
h1e = np.zeros((norb, norb))
np.fill_diagonal(h1e[:, 1:], -hopping)
np.fill_diagonal(h1e[1:, :], -hopping)
h1e[impurity_orb, impurity_orb + 1] = -hybridization
h1e[impurity_orb + 1, impurity_orb] = -hybridization
h1e[impurity_orb, impurity_orb] = chemical_potential
# Two body matrix elements in the "position" basis
h2e = np.zeros((norb, norb, norb, norb))
h2e[impurity_orb, impurity_orb, impurity_orb, impurity_orb] = onsite
return h1e, h2e
def momentum_basis(norb: int) -> np.ndarray:
"""Get the orbital rotation to change from the position to the momentum basis."""
n_bath = norb - 1
# Orbital rotation that diagonalizes the bath (non-interacting system)
hopping_matrix = np.zeros((n_bath, n_bath))
np.fill_diagonal(hopping_matrix[:, 1:], -1)
np.fill_diagonal(hopping_matrix[1:, :], -1)
_, vecs = np.linalg.eigh(hopping_matrix)
# Expand to include impurity
orbital_rotation = np.zeros((norb, norb))
# Impurity is on the first site
orbital_rotation[0, 0] = 1
orbital_rotation[1:, 1:] = vecs
# Move the impurity to the center
new_index = n_bath // 2
perm = np.r_[1 : (new_index + 1), 0, (new_index + 1) : norb]
orbital_rotation = orbital_rotation[:, perm]
return orbital_rotation
def rotated(
h1e: np.ndarray, h2e: np.ndarray, orbital_rotation: np.ndarray
) -> tuple[np.ndarray, np.ndarray]:
"""Rotate the orbital basis of a Hamiltonian."""
h1e_rotated = np.einsum(
"ab,Aa,Bb->AB",
h1e,
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
h2e_rotated = np.einsum(
"abcd,Aa,Bb,Cc,Dd->ABCD",
h2e,
orbital_rotation,
orbital_rotation.conj(),
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
return h1e_rotated, h2e_rotated
# Total number of spatial orbitals, including the bath sites and the impurity
# This should be an even number
norb = 8
# System is half-filled
nelec = (norb // 2, norb // 2)
# One orbital is the impurity, the rest are bath sites
n_bath = norb - 1
# Hamiltonian parameters
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian in position basis
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
# Rotate to momentum basis
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
# In the momentum basis, the impurity is placed in the center
impurity_index = n_bath // 2
# Use PySCF to compute the exact ground state energy
reference_energy, _ = pyscf.fci.direct_spin1.kernel(h1e, h2e, norb, nelec)
A continuación, generamos los circuitos para producir los estados base de Krylov. Para cada especie de espín, el estado inicial está dado por la superposición de todas las excitaciones posibles de los tres electrones más cercanos al nivel de Fermi hacia los 4 modos vacíos más cercanos, partiendo del estado , y se realiza mediante la aplicación de siete XXPlusYYGates. Los estados evolucionados en el tiempo se producen mediante aplicaciones sucesivas de un paso de Trotter de segundo orden.
Para una descripción más detallada de este modelo y cómo se diseñan los circuitos, consulta "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization".
from typing import Sequence
import ffsim
import scipy
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit import CircuitInstruction, Qubit
from qiskit.circuit.library import CPhaseGate, XGate, XXPlusYYGate
def prepare_initial_state(qubits: Sequence[Qubit], norb: int, nocc: int):
"""Prepare initial state."""
assert norb >= 8
x_gate = XGate()
rot = XXPlusYYGate(0.5 * np.pi, -0.5 * np.pi)
for i in range(nocc):
yield CircuitInstruction(x_gate, [qubits[i]])
yield CircuitInstruction(x_gate, [qubits[norb + i]])
for i in range(3):
for j in range(nocc - i - 1, nocc + i, 2):
yield CircuitInstruction(rot, [qubits[j], qubits[j + 1]])
yield CircuitInstruction(
rot, [qubits[norb + j], qubits[norb + j + 1]]
)
yield CircuitInstruction(rot, [qubits[j + 1], qubits[j + 2]])
yield CircuitInstruction(
rot, [qubits[norb + j + 1], qubits[norb + j + 2]]
)
def trotter_step(
qubits: Sequence[Qubit],
time_step: float,
one_body_evolution: np.ndarray,
h2e: np.ndarray,
impurity_index: int,
norb: int,
):
"""A Trotter step."""
# Assume the two-body interaction is just the on-site interaction of the impurity
onsite = h2e[
impurity_index, impurity_index, impurity_index, impurity_index
]
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# One-body evolution for the full time
yield CircuitInstruction(
ffsim.qiskit.OrbitalRotationJW(norb, one_body_evolution), qubits
)
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# Time step
time_step = 0.2
# Number of Krylov basis states
krylov_dim = 8
# Initialize circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# Generate initial state
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
# Create list of circuits, starting with the initial state circuit
circuits = [circuit.copy()]
# Add time evolution circuits to the list
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
# Remove measurements
circuit.remove_final_measurements()
# Append another Trotter step
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
# Measure qubits
circuit.measure_all()
# Add a copy of the circuit to the list
circuits.append(circuit.copy())
circuits[0].draw("mpl", scale=0.4, fold=-1)
circuits[-1].draw("mpl", scale=0.4, fold=-1)
Paso 2: Optimizar el problema para la ejecución cuántica
A continuación, optimizamos el circuito para un hardware objetivo. Por ahora, crearemos un backend genérico con un número especificado de qubits y un conjunto de compuertas al que los circuitos de evolución temporal se descomponen de forma natural.
from qiskit.providers.fake_provider import GenericBackendV2
backend = GenericBackendV2(
2 * norb, basis_gates=["cp", "xx_plus_yy", "p", "x"]
)
Ahora, usamos Qiskit para transpilar los circuitos al backend objetivo.
from qiskit.transpiler import generate_preset_pass_manager
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
Paso 3: Ejecutar usando primitivas de Qiskit
Después de optimizar los circuitos para la ejecución en hardware, estamos listos para ejecutarlos en el hardware objetivo y recopilar muestras para la estimación de la energía del estado fundamental. Después de usar la primitiva Sampler para muestrear cadenas de bits de cada circuito, combinamos todos los resultados en un único diccionario de conteos y graficamos las 20 cadenas de bits muestreadas más comúnmente.
from qiskit.visualization import plot_histogram
from qiskit.primitives import StatevectorSampler
# Sample from the circuits
sampler = StatevectorSampler()
job = sampler.run(isa_circuits, shots=500)
from qiskit.primitives import BitArray
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
plot_histogram(bit_array.get_counts(), number_to_keep=20)
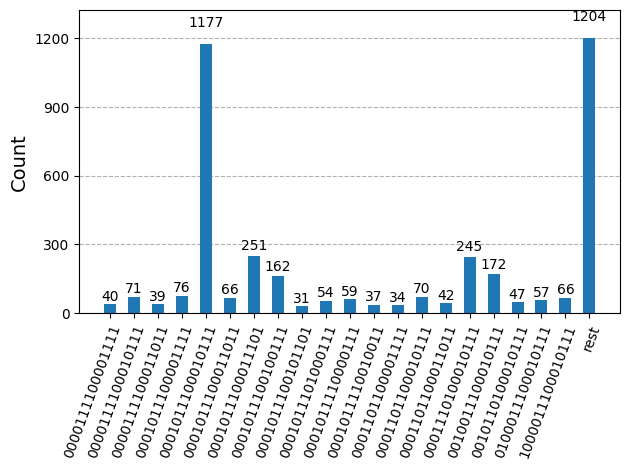
Paso 4: Posprocesar y devolver el resultado al formato clásico deseado
Ahora, ejecutamos el algoritmo SQD usando la función diagonalize_fermionic_hamiltonian. Consulta la documentación de la API para ver las explicaciones de los argumentos de esta función.
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -13.4222953188441
Subspace dimension: 529
Subsample 1
Energy: -13.42237556285828
Subspace dimension: 784
Subsample 2
Energy: -13.422045397387413
Subspace dimension: 529
Iteration 2
Subsample 0
Energy: -13.422379583305478
Subspace dimension: 900
Subsample 1
Energy: -13.422376197704326
Subspace dimension: 841
Subsample 2
Energy: -13.422421162849295
Subspace dimension: 1089
Iteration 3
Subsample 0
Energy: -13.422421164670345
Subspace dimension: 1156
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421205869572
Subspace dimension: 1156
Iteration 4
Subsample 0
Energy: -13.422421494558726
Subspace dimension: 1225
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421492737689
Subspace dimension: 1156
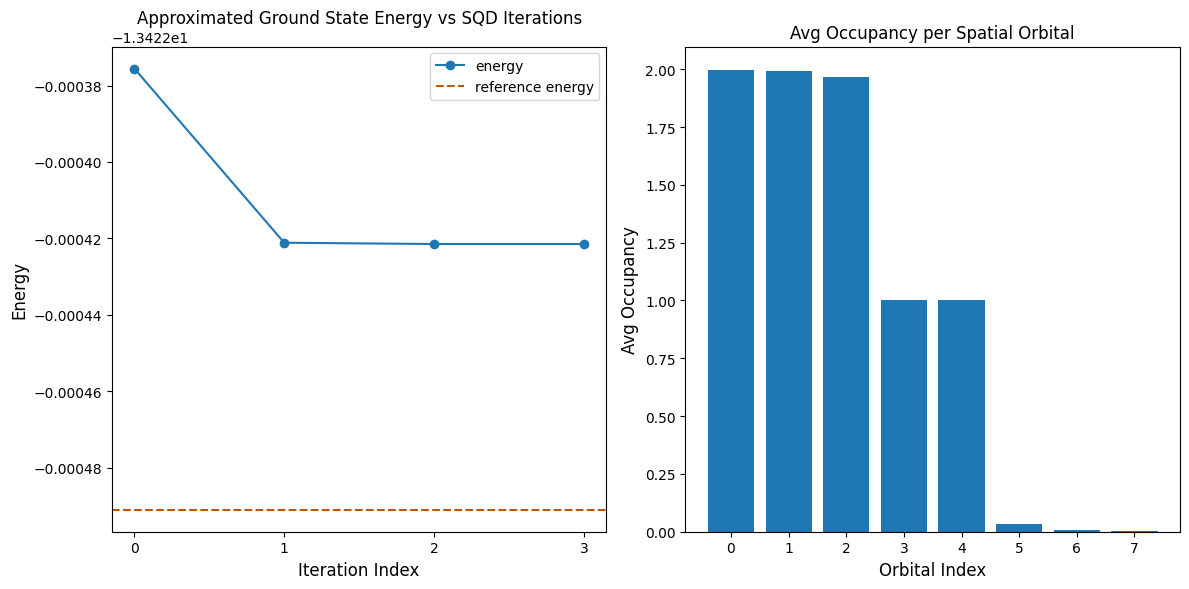
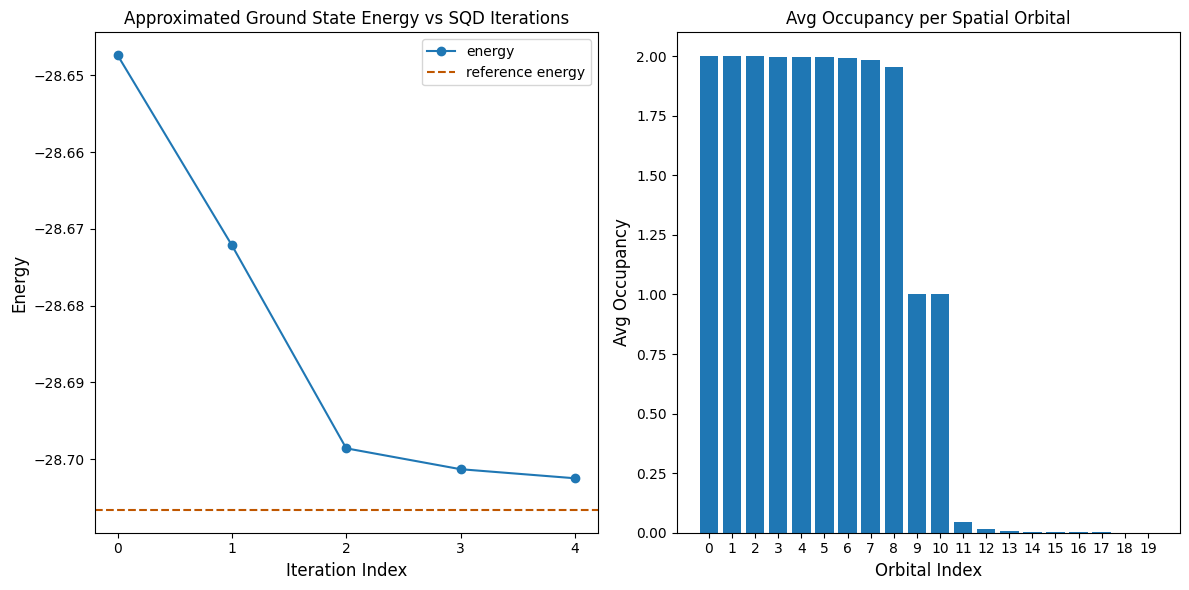
La siguiente celda de código grafica los resultados. El primer gráfico muestra la energía calculada en función del número de iteraciones de recuperación de configuración, y el segundo gráfico muestra la ocupación media de cada orbital espacial después de la iteración final. Dado que se trata de un problema tan pequeño, la primera iteración ya nos acerca mucho a la energía exacta (observa la escala del eje y).
import matplotlib.pyplot as plt
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
# Data for energies plot
x1 = range(len(result_history))
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Reference energy: -13.42249
SQD energy: -13.42242
Absolute error: 0.00007
Verificar la energía
La energía devuelta por SQD está garantizada como cota superior a la energía del verdadero estado fundamental. El valor de la energía puede verificarse porque SQD también devuelve los coeficientes del vector de estado que aproxima el estado fundamental. Puedes calcular la energía a partir del vector de estado usando sus matrices de densidad reducida de uno y dos partículas, como se demuestra en la siguiente celda de código.
rdm1 = result.sci_state.rdm(rank=1, spin_summed=True)
rdm2 = result.sci_state.rdm(rank=2, spin_summed=True)
energy = np.sum(h1e_momentum * rdm1) + 0.5 * np.sum(h2e_momentum * rdm2)
print(f"Recomputed energy: {energy:.5f}")
Recomputed energy: -13.42242
Ejemplo de hardware a gran escala
Ahora ejecutamos un ejemplo más grande en una QPU real. Para la energía de referencia, usamos los resultados de un cálculo DMRG que se realizó por separado.
from qiskit_ibm_runtime import SamplerV2 as Sampler
from qiskit_ibm_runtime import QiskitRuntimeService
# Model parameters
norb = 20
nelec = (norb // 2, norb // 2)
n_bath = norb - 1
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian and orbital rotation
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
impurity_index = n_bath // 2
# Set reference energy to DMRG value computed separately
reference_energy = -28.70659686
# Algorithm parameters
time_step = 0.2
krylov_dim = 8
# Construct circuits
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
circuits = [circuit.copy()]
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
circuit.remove_final_measurements()
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
circuit.measure_all()
circuits.append(circuit.copy())
# Initialize hardware backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=127
)
print(f"Using backend {backend.name}")
# Transpile to backend
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
# Sample from the circuits
sampler = Sampler(backend)
sampler.options.environment.job_tags = ["TUT_SKQD"]
job = sampler.run(isa_circuits, shots=500)
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
# Run configuration recovery and diagonalization
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
# Plot results
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
x1 = range(len(result_history))
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Using backend ibm_boston
Iteration 1
Subsample 0
Energy: -28.63965951544449
Subspace dimension: 9801
Subsample 1
Energy: -28.625588929202006
Subspace dimension: 9409
Subsample 2
Energy: -28.647371834135498
Subspace dimension: 8281
Iteration 2
Subsample 0
Energy: -28.67213260849567
Subspace dimension: 29584
Subsample 1
Energy: -28.670340686158816
Subspace dimension: 27225
Subsample 2
Energy: -28.669976379525988
Subspace dimension: 31329
Iteration 3
Subsample 0
Energy: -28.68622875601382
Subspace dimension: 36100
Subsample 1
Energy: -28.698569623143126
Subspace dimension: 34225
Subsample 2
Energy: -28.694848533971882
Subspace dimension: 33856
Iteration 4
Subsample 0
Energy: -28.69883392844593
Subspace dimension: 42025
Subsample 1
Energy: -28.701289495200996
Subspace dimension: 38025
Subsample 2
Energy: -28.699319594978245
Subspace dimension: 45369
Iteration 5
Subsample 0
Energy: -28.701936886834154
Subspace dimension: 51076
Subsample 1
Energy: -28.702468711812013
Subspace dimension: 53824
Subsample 2
Energy: -28.702298147575938
Subspace dimension: 52900
Reference energy: -28.70660
SQD energy: -28.70247
Absolute error: 0.00413
Próximos pasos
Si este trabajo te resultó interesante, puede que te interese el siguiente material:
- Diagonalización cuántica basada en muestras de un hamiltoniano químico - un tutorial relacionado que usa un ansatz variacional heurístico en lugar de circuitos de Trotter
- Diagonalización cuántica de Krylov de hamiltonianos de red - un tutorial sobre el método KQD
- Documentación de la API del complemento SQD - referencia para la función
diagonalize_fermionic_hamiltonian - Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization - el artículo en el que se basa este tutorial