HI-VQE Chemistry - Una Función de Qiskit por Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
Consulta la referencia de API
Las Funciones de Qiskit son una característica experimental disponible únicamente para los usuarios del Plan Premium, el Plan Flex y el Plan On-Prem (a través de la API de IBM Quantum Platform) de IBM Quantum®. Se encuentran en estado de versión preliminar y están sujetas a cambios.
Versiones de paquetes
El código de esta página se desarrolló utilizando los siguientes requisitos. Recomendamos usar estas versiones o más recientes.
qiskit-ibm-runtime~=0.45.0
Descripción general
En química cuántica, el problema de estructura electrónica se centra en encontrar las soluciones a la ecuación de Schrödinger electrónica: las funciones de onda cuánticas que describen el comportamiento de los electrones del sistema. Estas funciones de onda son vectores de amplitudes complejas, donde cada amplitud corresponde a la contribución de una posible configuración electrónica.
El estado fundamental es la función de onda de menor energía del sistema y tiene una importancia especial en el estudio de los sistemas moleculares. El enfoque más preciso para calcular el estado fundamental considera todas las configuraciones electrónicas posibles, pero esto se vuelve intratable para sistemas más grandes, ya que el número de configuraciones crece exponencialmente con el tamaño del sistema.
El Variational Quantum Eigensolver Iterativo de Handover (HI-VQE) es un innovador método híbrido cuántico-clásico para estimar con precisión el estado fundamental de sistemas moleculares. Integra hardware cuántico con computación clásica: usa procesadores cuánticos para explorar eficientemente configuraciones electrónicas candidatas y calcula la función de onda resultante en computadoras clásicas. Al generar funciones de onda compactas pero químicamente precisas, HI-VQE mejora la investigación y el descubrimiento en química cuántica y ciencia de materiales.

HI-VQE reduce la complejidad computacional del problema de estructura electrónica estimando eficientemente el estado fundamental con alta precisión. Se enfoca en un subconjunto cuidadosamente seleccionado de las configuraciones electrónicas más relevantes, optimizando tanto la precisión como la eficiencia.
Combinando las fortalezas de las computadoras clásicas y cuánticas, HI-VQE refina e itera de manera progresiva la estimación actual de la función de onda. Sus técnicas únicas de construcción de subespacios ayudan a hacer la selección de configuraciones más eficiente, de modo que los usuarios tengan mayor control computacional y mayor precisión en las simulaciones de química cuántica.
Si deseas aprender sobre el algoritmo con más profundidad, puedes leer el artículo de investigación asociado.
Descripción
El número de configuraciones electrónicas para un sistema molecular crece exponencialmente con el tamaño del sistema. Sin embargo, para ciertos estados electrónicos, como el estado fundamental, es común que solo una pequeña fracción de configuraciones contribuya de forma significativa a la energía del estado. Los métodos de interacción de configuraciones seleccionadas (SCI, por sus siglas en inglés) aprovechan esta escasez para reducir los costos computacionales identificando y enfocándose en las configuraciones más relevantes. Este subconjunto de configuraciones se denomina subespacio.
HI-VQE aprovecha la eficiencia inherente de las computadoras cuánticas para representar sistemas moleculares y asistir en la búsqueda de subespacios. Integra subrutinas clásicas y cuánticas para resolver el problema de estructura electrónica con alta precisión. A diferencia de los métodos cuánticos SCI existentes, HI-VQE combina entrenamiento variacional, construcción iterativa de subespacios y cribado de configuraciones por prediagonalización para mejorar la eficiencia reduciendo las mediciones cuánticas, las iteraciones y los costos de diagonalización clásica. HI-VQE puede aplicarse, por lo tanto, a sistemas moleculares más grandes que requieren más qubits, y reduce el costo de resolver un problema de un tamaño dado al mismo grado de precisión.
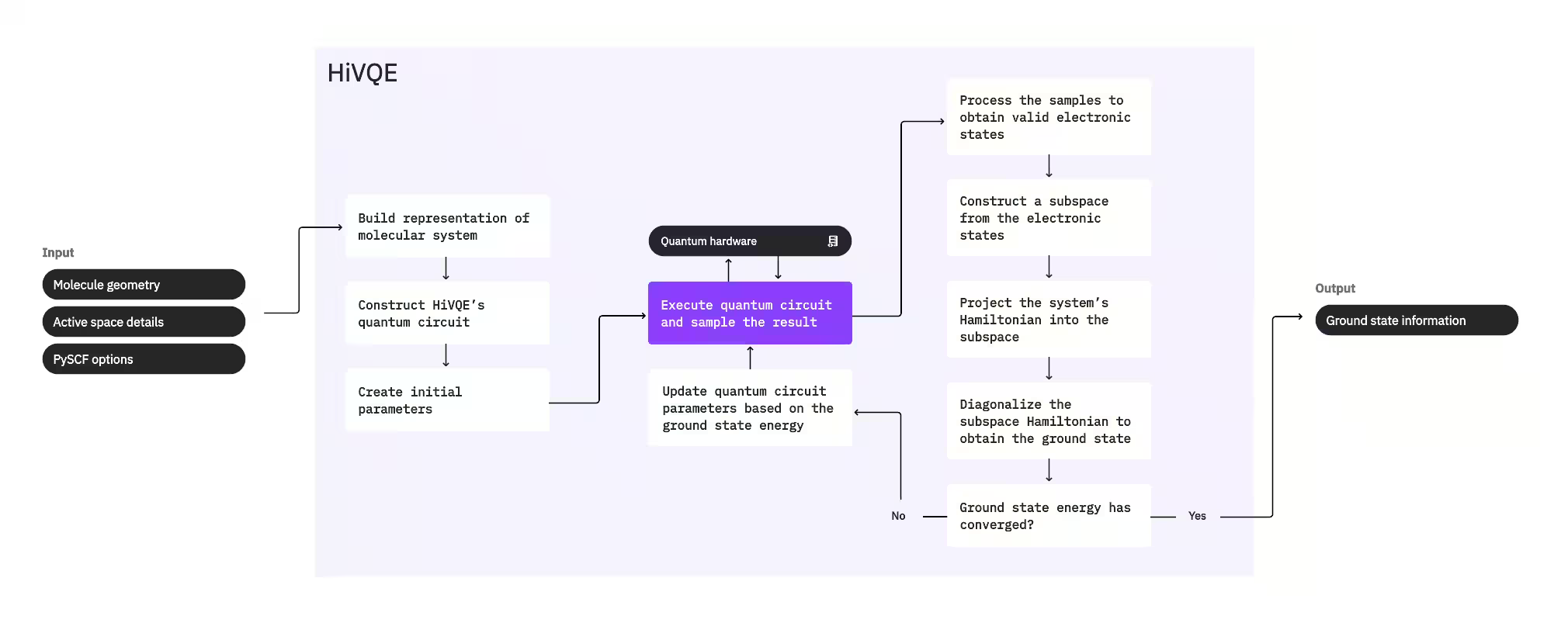
Para calcular el estado fundamental de un sistema, HI-VQE primero utiliza el paquete de química clásica PySCF para generar una representación molecular a partir de las entradas del usuario, como la geometría molecular y otra información molecular. Luego entra en un ciclo de optimización híbrido cuántico-clásico, refinando iterativamente un subespacio para representar de forma óptima el estado fundamental mientras minimiza el número de configuraciones incluidas. El ciclo continúa hasta que se cumplen los criterios de convergencia, como el tamaño del subespacio o la estabilidad energética, tras lo cual se obtienen como salida la función de onda del estado fundamental calculado y su energía. Estos resultados pueden usarse para construir superficies de energía potencial precisas y realizar análisis adicionales del sistema.
El ciclo de optimización se centra en ajustar los parámetros de un circuito cuántico para generar un subespacio de alta calidad. HI-VQE ofrece tres opciones de circuito cuántico: excitation_preserving, efficient_su2 y LUCJ. La optimización se inicializa cerca del estado de referencia Hartree-Fock por su adecuación general. Luego el circuito se ejecuta en un dispositivo cuántico y las configuraciones se muestrean del estado cuántico resultante antes de devolverse como cadenas binarias. Debido al ruido del dispositivo cuántico, algunas configuraciones muestreadas pueden ser físicamente inválidas, al no conservar el número de electrones o el espín. HI-VQE aborda esto usando el proceso de recuperación de configuraciones del paquete qiskit-addon-sqd, de modo que los usuarios pueden corregir las configuraciones inválidas o descartarlas.
Las configuraciones válidas pasan entonces por un paso de cribado opcional para eliminar aquellas que se predice que contribuirán mínimamente. Esto reduce la dimensión del subespacio, reduciendo así el costo del paso de diagonalización. Si el cribado está habilitado, se construye un Hamiltoniano de subespacio preliminar a partir de las configuraciones válidas y se realiza una diagonalización con criterios de terminación muy laxos. Aunque la precisión de las amplitudes resultantes para cada configuración es baja, es eficaz para predecir qué configuraciones excluir del subespacio en esta iteración, y es rápido de calcular.
Las configuraciones seleccionadas se añaden al subespacio y el Hamiltoniano del sistema se proyecta en este subespacio. El subespacio se actualiza iterativamente, preservando las configuraciones más relevantes entre iteraciones. Este enfoque contrasta con métodos alternativos porque el circuito cuántico no necesita aproximar el estado fundamental completo en cada paso.
A continuación, el Hamiltoniano del subespacio se diagonaliza clásicamente para obtener el menor valor propio y su vector propio correspondiente, que representa una aproximación del estado fundamental y su energía. A medida que la calidad del subespacio mejora con las iteraciones, el estado fundamental calculado se aproxima mejor al verdadero estado fundamental. En este punto puede realizarse un paso de cribado adicional para eliminar del subespacio cualquier configuración que no tenga una contribución sustancial al estado fundamental calculado. Este paso garantiza que el subespacio llevado a la siguiente iteración sea lo más compacto posible. Esto se evalúa en función de las amplitudes que devuelve la diagonalización, ya que representan la contribución de importancia de cada configuración al estado fundamental calculado.
Una verificación de convergencia determina entonces si un entrenamiento adicional mejoraría los resultados. Si es así, se realiza un paso de expansión clásica opcional, los parámetros del circuito cuántico se actualizan para minimizar aún más la energía calculada y el proceso se repite. El paso de expansión clásica genera configuraciones adicionales para el subespacio, complementando las configuraciones muestreadas del dispositivo cuántico. Primero identifica la configuración con la mayor amplitud en los resultados de la diagonalización, antes de generar nuevas configuraciones con excitaciones simples y dobles a partir de la configuración identificada. El número deseado de estas configuraciones se añade entonces al subespacio.
Una vez que se determina que las iteraciones han convergido, HI-VQE devuelve el estado fundamental calculado (en forma de los estados del subespacio y sus amplitudes en la función de onda del estado fundamental), su energía y una medida de varianza de energía que indica si el estado calculado forma un estado propio del Hamiltoniano del sistema.
Los usuarios pueden decidir el circuito cuántico utilizado y el número de disparos por circuito cuántico, así como controlar el tamaño del subespacio o habilitar la generación clásica de configuraciones adicionales para asistir a las configuraciones generadas cuánticamente. De este modo, los usuarios pueden adaptar el comportamiento de HI-VQE a sus aplicaciones deseadas.
Licencias
Ten en cuenta que el uso de esta Función de Qiskit está limitado a problemas que requieren como máximo 20 qubits, a menos que se obtenga una licencia que otorgue un límite mayor.
Envía un correo electrónico a qiskit.support@qunovacomputing.com si deseas consultar sobre la obtención de una licencia.
Primeros pasos
Primero, solicita acceso a la función. Luego, autentícate con tu clave API de IBM Quantum® y, suponiendo que ya hayas guardado tu cuenta en tu entorno local, selecciona la Función de Qiskit de la siguiente manera:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
Ejemplo
El primer ejemplo muestra cómo calcular la energía del estado fundamental de una molécula de NH3 usando el algoritmo HI-VQE.
Definir la geometría molecular y las opciones
La geometría molecular del NH3 se proporciona con coordenadas cartesianas separadas por ";" para cada átomo.
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
Se pueden definir y proporcionar opciones adicionales para el sistema molecular en el siguiente formato de diccionario.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
Ejecuta la función con las entradas de geometría y opciones.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
Es buena idea imprimir el ID del trabajo de la Función para poder proporcionarlo en solicitudes de soporte si algo sale mal.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
Este ejemplo utiliza 16 qubits con 8 orbitales de base sto3g para una molécula de NH3. Verifica el estado de tu carga de trabajo de Función de Qiskit u obtén los resultados de la siguiente manera:
print(job.status())
QUEUED
Una vez completado el trabajo, los resultados pueden obtenerse con la instancia result().
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
Para acceder a la energía del estado fundamental, usa la clave "energy". La clave "eigenvector" proporciona los coeficientes CI con la notación de cadena de bits correspondiente de la configuración electrónica almacenada con "states" de los resultados.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
Rendimiento
Esta sección muestra los cálculos de referencia demostrados de HI-VQE con un caso de 24 qubits para Li2S, un caso de 40 qubits para una molécula de N2 y un caso de 44 qubits para un sistema FeP-NO.
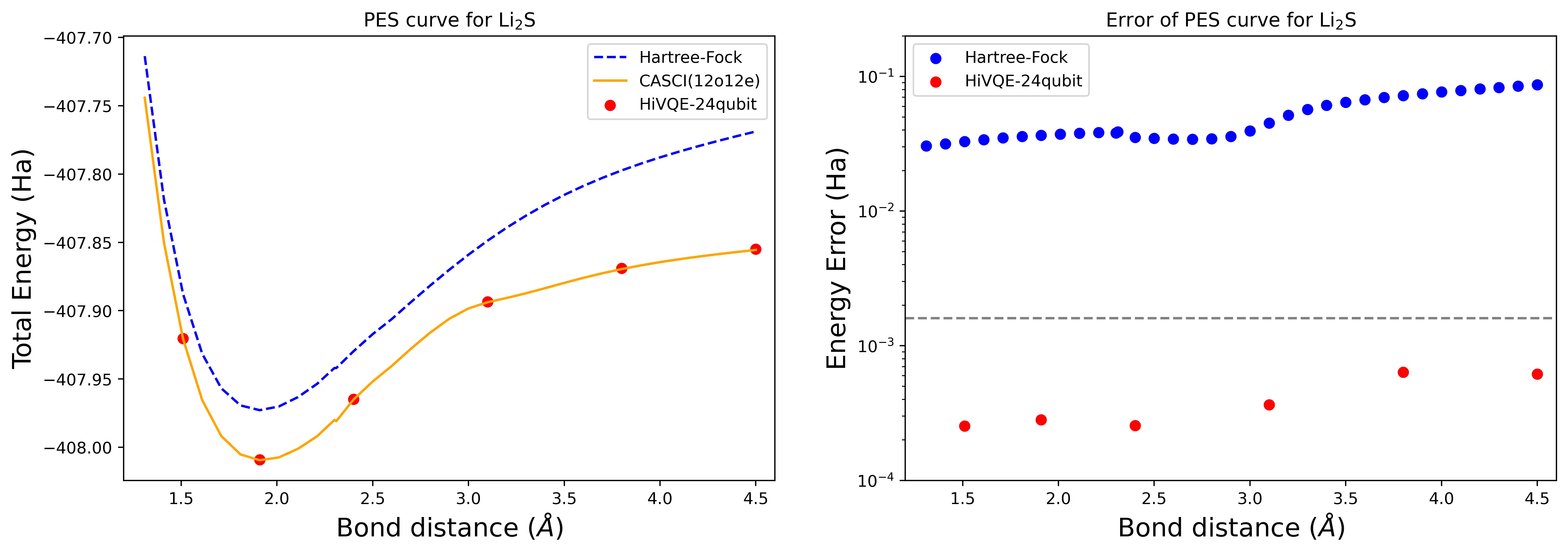
Curva de superficie de energía potencial de disociación para una molécula de Li2S con 24 qubits
La curva PES se muestra con la referencia FCI y la estimación inicial de RHF, junto con el error de energía respecto a la referencia FCI.
 .
.
Los cálculos se han realizado con las siguientes geometrías y opciones.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
Los puntos rojos representan los resultados del cálculo HI-VQE para seis geometrías diferentes, y tres geometrías correspondientes a 1.51, 2.40 y 3.80 Angstrom se proporcionan como entrada en la celda anterior.
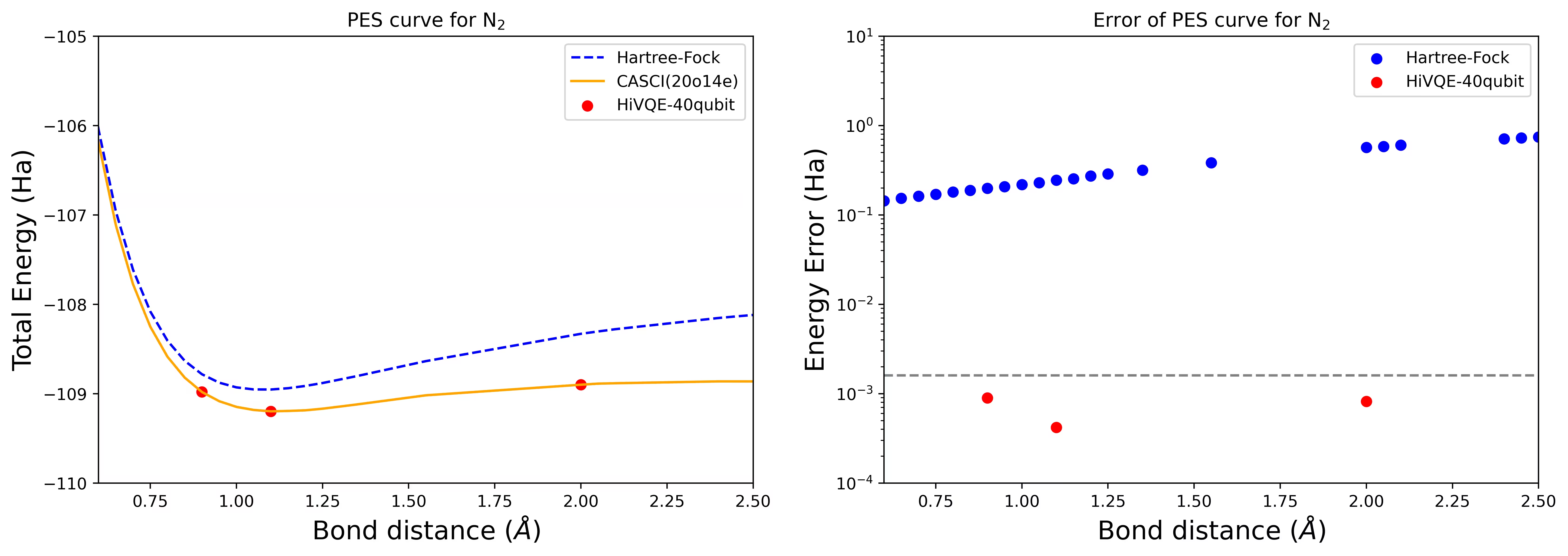
Curva PES de disociación para una molécula de N2 con 40 qubits
La molécula de nitrógeno ha sido identificada como un sistema multirreferencial con grandes contribuciones de energía de correlación más allá del estado Hartree-Fock. Realizamos un cálculo de referencia para la molécula de N2 con base cc-pvdz, (20o,14e) usando la selección de orbitales activos homo-lumo. El número del espacio activo completo (CAS) para representar este problema es 6 009 350 400. No es posible obtener la solución del problema de valores propios (para la energía y la estructura electrónica) con este número de estados usando una computadora de escritorio potente (16 cpu/64 GB). Con HI-VQE, los usuarios pueden buscar eficientemente el subespacio de estados CAS para encontrar resultados con precisión química mientras ahorran recursos computacionales de forma significativa. Los siguientes gráficos muestran la curva PES del cálculo HI-VQE de 40 qubits de la disociación de la molécula de N2.
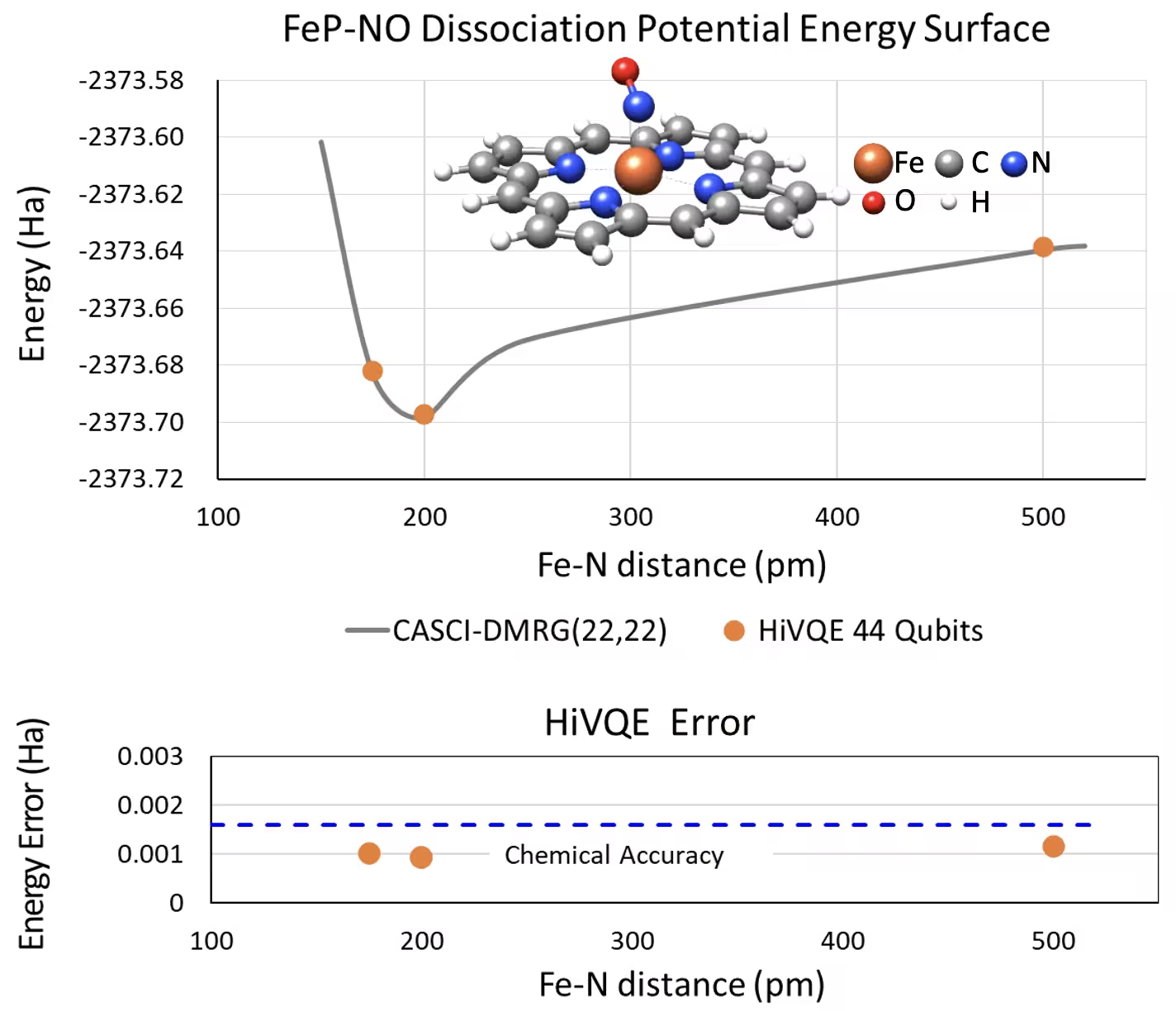
Curva PES de disociación para el hierro(II)-porfirina pentacoordinado con un sistema NO con 44 qubits
Otro sistema químico interesante es un complejo de hierro(II)-porfirina (FeP) con un ligando de óxido nítrico (NO) coordinado, que representa un sistema de metaloportirina biológicamente relevante que desempeña roles cruciales en diversos procesos fisiológicos. En este ejemplo, se ha utilizado HI-VQE para estimar la curva precisa de superficie de energía potencial de la interacción intermolecular entre FeP y NO (energía del estado fundamental para geometrías con diferentes separaciones). El sistema combinado tiene 450 orbitales y 202 electrones (450o,202e) con base 6-31g(d) en total. La selección de orbitales activos homo-lumo se utilizó para calcular el caso más pequeño del caso real con (22o,22e). A partir de los siguientes resultados de referencia, pudimos lograr la precisión química (> 1.6 mHa) con un cálculo de química clásica de vanguardia de CASCI(DMRG) (22o,22e) como referencia.
Puntos de referencia
- El tamaño exacto de la matriz es el número de determinantes para la solución exacta, como FCI y CASCI.
- El cálculo de HI-VQE muestrea y calcula el subespacio de este (es decir, el tamaño de la matriz HI-VQE).
- El tiempo total incluye el tiempo de ejecución en QPU y las ejecuciones de la Función de Qiskit con CPU.
- La precisión se estima a partir de la diferencia de energía respecto a la solución exacta.
| Sistema químico | Número de qubits | Tamaño exacto de la matriz | Tamaño de la matriz HI-VQE | E(diff) de la exacta (mHa) | Número de iteraciones | Tiempo total | Uso del tiempo de QPU |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0.08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0.60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0.90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1.10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0.90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1.20 | 21 | 46547 s | 258 s |
Obtener mensajes de error
Si tu carga de trabajo falla, el estado será ERROR y llamar a job.result() generará una excepción:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
Obtener soporte
Puedes enviar un correo electrónico a qiskit.support@qunovacomputing.com para obtener ayuda con esta función.
Si deseas ayuda para solucionar un error específico, proporciona el ID del trabajo de la Función correspondiente al trabajo que encontró el error.
Próximos pasos
- Solicita acceso a la función completando este formulario.
- Visita la referencia de API de esta Función de Qiskit.
- Prueba el tutorial Compute dissociation PES curve for FeP-NO with HI-VQE.
- Revisa Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- Prueba el tutorial Dissociation PES curves with Qunova HiVQE.