Mapeo (Mapping)
Mira el video sobre mapeo de Olivia Lanes, o ábrelo en una ventana separada en YouTube.
Introducción
En esta lección nos centraremos en el primer y a menudo más difícil paso para definir un programa cuántico: mapear un problema a un computador cuántico. Este paso describe cómo un usuario comienza con un problema computacional y lo traduce en algo que puede resolverse en un computador cuántico.
En las lecciones 2 y 3 de este curso, mencionamos que la fase de mapeo es el primero de cuatro pasos generales en el marco de Qiskit Patterns. De esas lecciones, quizás recuerdes que el objetivo del mapeo es traducir o reescribir un problema computacional en una función de costo o un valor esperado que podamos evaluar con un computador cuántico.
En la lección 3, discutimos un ejemplo concreto con Max-Cut, un problema computacionalmente difícil pero muy común en la optimización combinatoria. En ese ejemplo, pasamos por varios pasos para traducir el problema inicial del grafo en uno que pueda resolverse en un computador cuántico. Transformamos el problema de encontrar el número máximo de cortes en el grafo en una función de costo, reescribimos esa función de costo como un hamiltoniano y luego preparamos un estado cuántico de prueba cuyo estado fundamental corresponde al corte máximo. Finalmente, construimos un circuito cuántico que representa el estado de prueba de interés y luego añadimos las puertas específicas para permitir la evolución temporal del estado. Esta secuencia de pasos fue toda parte del mapeo. Mientras que los pasos exactos eran únicos para el problema Max-Cut, el mismo procedimiento general puede aplicarse a muchas otras aplicaciones, como la química cuántica y las simulaciones cuánticas.
El mapeo puede ser difícil. No hay una estrategia única para cada problema individual, por lo que puede ser intimidante. En esta lección, veremos algunas consideraciones generales para el mapeo y luego profundizaremos en algunos problemas de ejemplo representativos para demostrar las diferentes formas de mapear un problema a un computador cuántico.
Consideraciones generales
Mientras que la estrategia exacta que se utiliza para mapear un problema a un computador cuántico depende del problema en cuestión, en general cumplen con algunas cosas principales.
Primero, es posible que necesites simplificar el problema para hacerlo factible. Esto no es exclusivo de la computación cuántica -- todas las disciplinas científicas utilizan modelos simplificados para estudiar fenómenos que les interesan mientras ignoran detalles irrelevantes. En la física, hay una expresión famosa que lleva este principio al extremo: "Supongamos que la vaca es una esfera." A menudo es demasiado difícil describir un sistema exactamente como aparece, pero en su lugar podemos hacer simplificaciones razonables que aún pueden conducir a soluciones útiles. Algunos ejemplos de cómo podríamos hacer esto en la computación cuántica son limitar el tamaño o la profundidad del circuito eligiendo un ansatz eficiente en hardware, truncar evoluciones temporales complejas o despreciar términos del hamiltoniano que contribuyen poco a la energía final de un estado cuántico.
Segundo, el mapeo implica escribir el problema de una manera que un computador cuántico pueda entender. Esto a menudo implica hacerse estas tres preguntas:
- ¿Qué representarán nuestros qubits en nuestro modelo?
- ¿Es continuo nuestro problema? Dado que los computadores cuánticos son digitales, si el problema es continuo, necesitamos encontrar una forma de discretizarlo.
- Finalmente: ¿Coincide la topología del problema con la topología del hardware? Si no, es posible que necesitemos implementar algunos trucos para que funcione.
Abordemos la primera pregunta, que está en el centro de muchas dificultades al entender el mapeo: ¿Qué puede representar un qubit?
Los qubits pueden usarse para representar muchas cosas. Lo primero y quizás lo más simple es un nodo en un grafo. Los grafos se utilizan para mostrar la conectividad en muchos tipos diferentes de problemas matemáticos, y los nodos son un elemento fundamental que representa un punto o entidad en una red. Dependiendo de lo que represente la red completa, eso podría ser una ciudad, una persona o un ferromagneto en una red.
Los qubits también pueden usarse para representar bosones y fermiones, aunque debo advertir aquí que un solo qubit no corresponde exactamente a un bosón o un fermión -- es algo más complicado que eso, como discutiremos más adelante en la lección.
Ahora los ejemplos se vuelven un poco más complicados. Con estos modelos, ya no tiene sentido hablar de qubits individuales; en su lugar, necesitamos grupos de ellos para producir algo físico. Por ejemplo, un grupo de qubits, aquí representados en una topología Heavy-Hex, puede usarse para representar posiciones geométricas de aminoácidos; cadenas de polímeros. Otro ejemplo es la simulación de dispersión de hadrones en modelos de física de altas energías, que puede hacerse simulando la evolución temporal de un paquete de ondas hadrónico. En este caso, un registro de qubits puede usarse para codificar diferentes estados de un campo cuántico; el estado de vacío de ese campo o un paquete de ondas propagándose sobre ese vacío.
Pero a este punto hemos hablado de manera suficientemente abstracta sobre el desafío que tenemos por delante. Veamos estos ejemplos en detalle.
Ejemplos de mapeo
Max-Cut
Comencemos con nuestro primer ejemplo. Uno de los problemas de mapeo más simples es el que ya hemos tratado en profundidad: el ejemplo de Max-Cut. En ese problema, el mapeo fue relativamente fácil para nosotros, ya que un qubit correspondía a un nodo en nuestro grafo.
Recuerda: para mapear el problema de Max-Cut, expresamos la función de costo como un hamiltoniano usando la formulación QUBO. Una función de costo hamiltoniana es una función que codifica la solución óptima del problema en el estado fundamental del hamiltoniano. Para construir el hamiltoniano de costo, usamos la clase SparsePauliOp en Qiskit para especificar la conectividad de nuestro grafo, y la fase de mapeo a los operadores cuánticos estaba completa. Y el circuito cuántico era simplemente el ansatz QAOA. Como repaso, consulta la lección Utility-Scale QAOA, donde repasamos todo esto con mucho más detalle.
En esa lección, la conectividad del grafo en el ejemplo de 100 qubits a escala de utilidad ya coincidía con la topología de nuestro hardware superconductor. Así que no tuvimos que preocuparnos por cómo manejar diferentes topologías. Pero eso no siempre es el caso. Si tuviéramos un grafo más complicado o más densamente conectado que los ejemplos destacados hasta ahora, necesitaríamos implementar una serie de puertas SWAP para cambiar la conectividad efectiva del hardware. Esto se maneja en el segundo paso de Qiskit Patterns, la transpilación, pero debería tenerse en mente ya en el paso de mapeo.
Plegamiento de proteínas
A continuación, veamos un ejemplo que fue modelado en el artículo "Resource-efficient quantum algorithm for protein folding", escrito por IBM® y colaboradores de la University of New South Wales.
Un poco de contexto bioquímico: las proteínas son macromoléculas compuestas por largas cadenas de aminoácidos. Estas cadenas se pliegan en estructuras complejas que realizan una variedad de funciones biológicas. La determinación de la estructura proteica en el espacio tridimensional y la comprensión de las relaciones entre estructura y función están entre los problemas más desafiantes en la bioquímica actual. Las proteínas se pliegan en estructuras útiles debido a las interacciones entre aminoácidos. Cuando una estructura gira y se pliega, aminoácidos que están muy separados a lo largo de la cadena pueden quedar directamente uno al lado del otro e interactuar fuertemente.
Para modelar esto en un computador cuántico, necesitamos un hamiltoniano que describa todas estas interacciones entre aminoácidos. Luego podemos predecir la estructura final encontrando el estado que minimiza la energía de nuestro hamiltoniano. Aquí nos centraremos en cómo las cadenas de aminoácidos pueden modelarse en un computador cuántico y cómo podemos obtener distancias inter-aminoácidos para calcular las energías de interacción. Con eso tendremos todas las contribuciones necesarias al hamiltoniano para simularlo en un computador cuántico.
En las proteínas reales, los aminoácidos pueden ocupar un continuo de posiciones posibles. Sin embargo, usaremos una simplificación y los restringiremos con un modelo de red, donde cada aminoácido ocupa un punto en una red. Aquí, los autores usaron una red tetraédrica. Nota breve: aquí codificamos la dirección de las aristas, no los nodos mismos como en el problema Max-Cut. Cada qubit representa un posible camino de un solo paso a lo largo de la red tetraédrica. Observa que los sitios de red adyacentes se han etiquetado como A o B, debido a sus diferentes orientaciones en la red.
La cadena de proteínas se representa como una serie de giros o direcciones en esta red. Cada giro entre aminoácidos puede ir en una de cuatro direcciones, correspondientes a las aristas del tetraedro. Estos cuatro giros posibles se codifican con cuatro qubits en los estados 0001, 0010, 0100 o 1000.
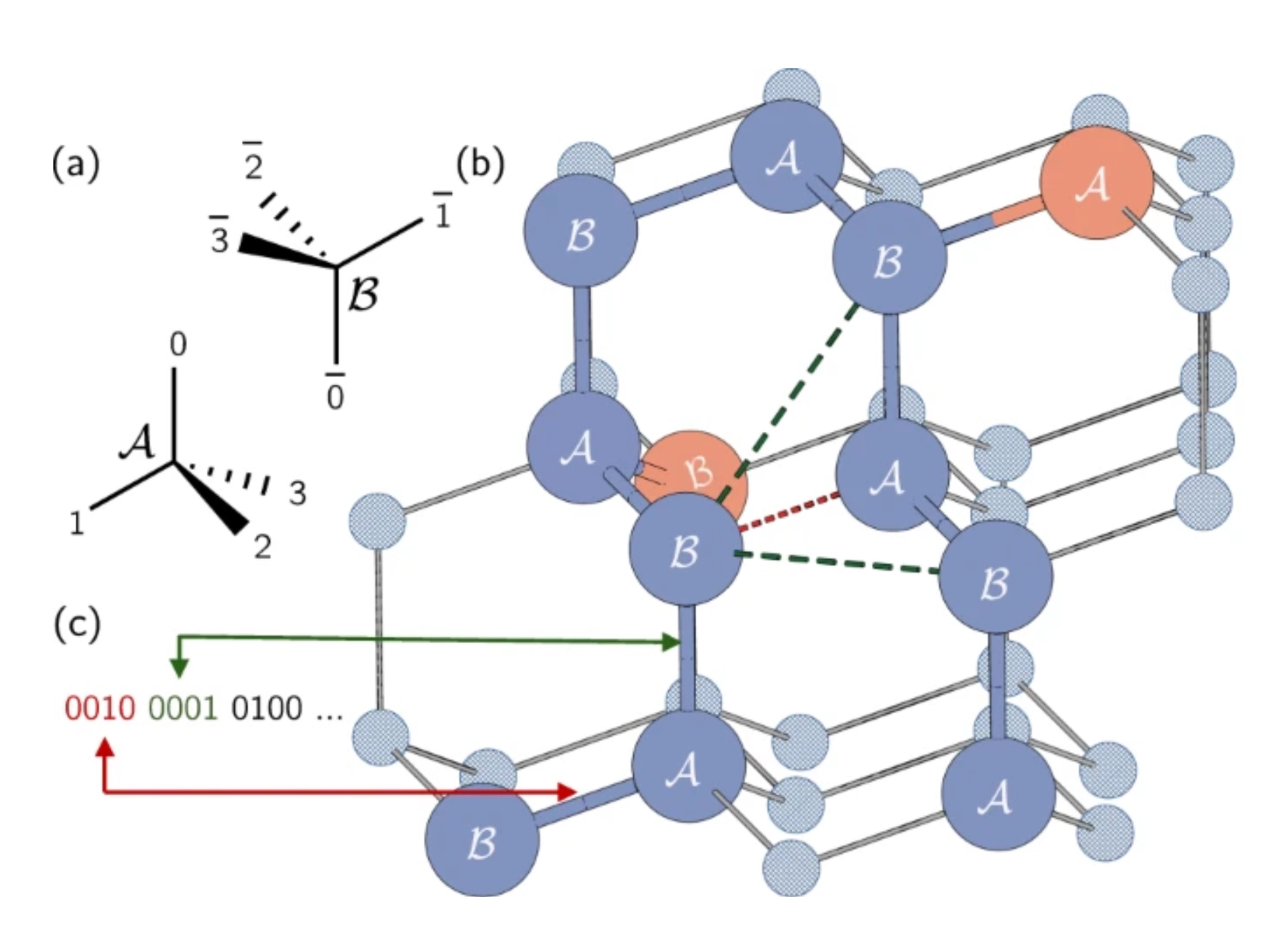
Veamos un ejemplo en la figura de arriba. Coloquemos nuestro primer aminoácido en el punto etiquetado "B", que está circulado en rojo en nuestra red tetraédrica. La dirección del primer aminoácido al segundo es arbitraria, ya que el sistema siempre puede rotarse para que esta arista apunte en cualquier dirección. Así podemos colocar nuestro segundo aminoácido en el punto debajo del primero, etiquetado "A". No es fácil de ver, pero el camino del segundo al tercero también es arbitrario. Las tres opciones resultarían en tener dos aristas con un ángulo de aproximadamente 109,5 grados entre ellas. La selección de esta segunda arista simplemente determina la orientación de nuestra proteína en el espacio. Sin pérdida de generalidad, podemos elegir los dos primeros giros simplemente como 0001 y 0010.
Con estas simplificaciones, la configuración de la cadena de aminoácidos viene dada por esta expresión:
Hasta ahora hemos mapeado las aristas del tetraedro a qubits, y nuestro circuito cuántico será un ansatz. Ahora necesitamos considerar cómo codificamos la energía del problema en un hamiltoniano, de modo que su estado fundamental nos dé el patrón de plegamiento óptimo.
Para cada configuración dada, hay una energía asociada debido a las interacciones entre los aminoácidos. Estas interacciones son más fuertes cuando los dos aminoácidos están cerca uno del otro. Obviamente, los aminoácidos adyacentes en la columna vertebral de la cadena siempre interactuarán entre sí. Pero debido a que la proteína puede girar y plegarse, otros pares de aminoácidos también pueden interactuar. Los aminoácidos 10 y 20 podrían, por ejemplo, terminar en sitios adyacentes después de que la proteína se pliega. Así que necesitamos una fórmula para describir la distancia entre los aminoácidos y usando la información codificada en los qubits de configuración. De esta manera, podemos usar su distancia de separación para determinar cuán fuertemente interactúan.
Primero, introducimos una función que indica si una arista se usa para el giro en el aminoácido o no. Aquí puede tomar una de las cuatro direcciones posibles. Basándonos en la configuración con la que comenzamos arriba, podemos escribir estas funciones:
Luego podemos definir una diferencia en el número de giros marcados con en las redes A y B desde el índice hasta el índice como :
¿Por qué haríamos esto? Bueno, resulta que un giro de del sitio de red A a B se cancela exactamente con un giro de del sitio de red B a A. Así que para conocer la distancia del aminoácido en la posición al de la posición , solo necesitamos encontrar la diferencia entre los pasos en aristas desde sitios A y sitios B. Como los sitios A y B necesariamente se alternan a lo largo de la columna vertebral de la proteína, esto se captura por el . El mismo argumento se aplica para los cuatro tipos de aristas. Así, la distancia total entre aminoácidos en pasos de red tetraédrica puede calcularse mediante esta expresión:
Pero, ¿cómo obtenemos el hamiltoniano a partir de esta larga ecuación para la distancia total entre los aminoácidos? Primero, podemos convertir de distancia en pasos de red al espacio euclidiano con geometría simple:
Estas distancias se usan luego para calcular la energía de la configuración proteica. Dependiendo de nuestros propósitos, podríamos establecer una distancia límite por debajo de la cual consideramos que el par interactúa, o podríamos hacer algo más complicado.
Puede que no sea obvio, pero en realidad hemos terminado con la fase de mapeo al hacer esto. Los estados de los qubits indican el "giro" de la proteína en cada punto de la red, y la colección de giros determina la distancia entre cada par de aminoácidos. Pares de diferentes tipos de aminoácidos tienen diferentes interacciones, algunas atractivas, otras repulsivas. Si usas la configuración y las distancias solo para determinar si las interacciones conocidas entre aminoácidos están "activadas" o "desactivadas", las intensidades de estas interacciones ya han sido elaboradas y pueden consultarse simplemente en una tabla como esta:
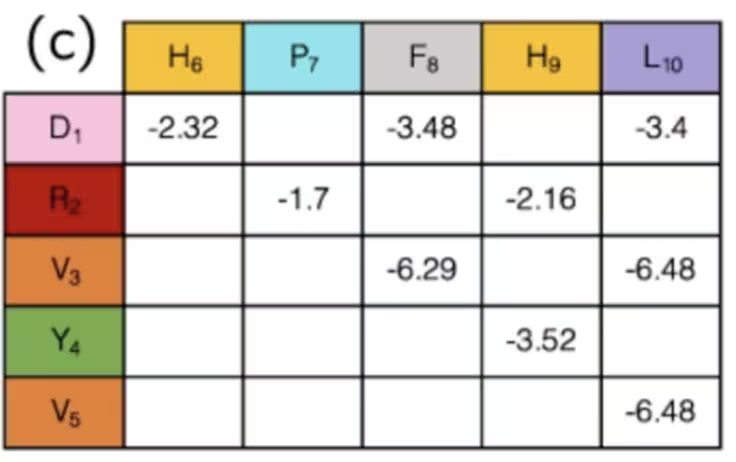
En resumen: en este ejemplo, los qubits se usan para marcar pasos en un camino a lo largo de una red, que juntos forman una cadena de aminoácidos. Al simular cómo se dobla y pliega, podemos esperar obtener mejores resultados en la investigación médica. Omitimos cómo calcular algunos términos de este hamiltoniano, ya que eran hiperespecíficos para este problema, mientras que definir direcciones en una red puede aplicarse más generalmente. Una vez que tienes un hamiltoniano general, siempre querrás traducirlo a operadores de Pauli, que son nativos del computador cuántico. Eso es lo que discutiremos a continuación.
Transformación de Jordan-Wigner
Ahora exploremos cómo traducir un sistema de partículas subatómicas a operadores de Pauli.
Las partículas subatómicas se dividen en dos categorías -- bosones y fermiones. Los bosones, como los fotones o el Higgs, obedecen un conjunto particular de reglas estadísticas. Los fermiones, como los electrones o los neutrinos, obedecen otro. La diferencia clave entre ellos es que los bosones pueden ocupar el mismo estado -- no hay límite para cuántos bosones pueden estar en el estado fundamental o en un estado excitado. Los fermiones, por otro lado, son "egoístas" -- exigen que cada partícula individual tenga su propio estado cuántico.
Los bosones también tienen espines enteros, mientras que los fermiones tienen espines semienteros, como el electrón de espín 1/2 y las partículas más exóticas de espín 3/2. Para describir un sistema de partículas, necesitamos una descripción de su energía. Centrémonos en los fermiones. Podemos comenzar con un hamiltoniano escrito en términos de lo que llamamos operadores c. Estos son básicamente objetos matemáticos que corresponden a crear o destruir un fermión de un estado en el sistema. A menudo se escriben como y , donde es el operador que crea un fermión en el estado , y es el operador que destruye un fermión en el estado .
Pero recuerda: los computadores cuánticos normalmente operan en una base de qubits con reglas específicas para representar sistemas fermiónicos; no operan inherentemente en el lenguaje de operadores fermiónicos. Para cerrar esta brecha, necesitamos mapear esta notación fermiónica a operadores de Pauli, que corresponden naturalmente a puertas cuánticas.
Hay varias transformaciones de este tipo para fermiones. Una opción común es la transformación de Jordan-Wigner. Las codificaciones de Bravyi-Kitaev y de paridad son codificaciones fermiónicas más recientes. Los operadores bosónicos pueden transformarse mediante la transformación de Holstein-Primakoff o mediante el mapeo directo de los estados de Fock a una subbasis de los modos bosónicos disponibles. Otras codificaciones también están siendo activamente investigadas. Por ahora, nos centraremos únicamente en la transformación de Jordan-Wigner.
La transformación de Jordan-Wigner implica el mapeo de un solo fermión a muchos qubits. ¿Por qué no podemos simplemente asignar un qubit para representar cada electrón? Esto tiene que ver con la distinguibilidad de fermiones idénticos. Los qubits son distinguibles y los electrones no. Por ejemplo, podemos etiquetar e identificar fácilmente qubits individuales en cualquier dispositivo. Pero la indistinguibilidad de los electrones significa que no podemos etiquetarlos en absoluto. Por lo tanto, debemos etiquetar los operadores según los orbitales ocupados, como 1s, 2p, 2p, etc., en lugar de electrones específicos. Cada qubit juega aproximadamente el papel de un orbital en la molécula, que está ocupado o desocupado. Pero cómo hacemos esto es algo más complicado. El mapeo de Jordan-Wigner rastrea la antisimetría y asegura las estadísticas correctas de todo el sistema fermiónico. El mapeo de Jordan-Wigner expresa los operadores fermiónicos en términos de operadores de Pauli con estas relaciones:
El mapeo de Jordan-Wigner es conceptualmente simple debido a la correspondencia uno a uno entre orbitales y qubits. Hay otros mapeos que logran un objetivo similar, incluyendo el mapeo de paridad. El cálculo de la paridad de un estado requiere considerar múltiples qubits. En el mapeo de paridad (y algunos otros), la interpretación de que un qubit corresponde a un orbital no se aplica. Ahora repasemos un ejemplo sencillo. Supongamos que queremos calcular la interacción de un solo qubit . Comenzamos sustituyendo nuestras definiciones para los operadores de creación y destrucción.
El conmutador . Así que la expresión final se convierte en:
Así que hemos reescrito exitosamente una expresión fermiónica en operadores de Pauli que nuestro computador cuántico puede entender.
Discutamos brevemente cómo implementaríamos el mapeo de Jordan-Wigner en Qiskit. Es importante entender cómo funcionan este tipo de transformaciones, pero sería impráctico ejecutarlas manualmente cada vez -- especialmente para sistemas grandes. Afortunadamente, Qiskit nos lo facilita con la función SparsePauliOp.
A un alto nivel, los pasos son:
- Usa la función
from_listde SparsePauliOp para crear un operador identidad que coincida con el tamaño del espacio de parámetros a mapear. - Según la definición de los operadores de creación y destrucción mostrada anteriormente, usa la función
from_listde SparsePauliOp para definir operadores de Pauli , , . Esto mapeará los operadores fermiónicos de creación y destrucción a operadores de espín de qubit y codificará el número de ocupación fermiónico en la base computacional de los qubits. - Genera el hamiltoniano deseado en la base de Pauli aplicando los operadores anteriores a los orbitales de interés. Esto normalmente corresponde a crear una matriz identidad que represente los orbitales centrales (no interactuantes), y luego aplicar los operadores de creación y destrucción al espacio activo con pesos que correspondan a las particularidades del problema en cuestión.
Ahora que entendemos completamente el esquema de mapeo de Jordan-Wigner, veamos un ejemplo más complicado. Quizás recuerdes el artículo titulado "Scalable Circuits for Preparing Ground States on Digital Quantum Computers: The Schwinger Model Vacuum on 100 Qubits" de la lección anterior. No repasaremos el artículo en detalle de nuevo -- solo nos centraremos en el mapeo de Jordan-Wigner que se usa para expresar el hamiltoniano de los puntos de red con sitios para el modelo de Schwinger en ausencia de un campo eléctrico.
Aquí es mucho más difícil determinar exactamente qué representa un qubit en este modelo, porque solo es la colección de qubits juntos lo que produce algo físico, en este caso un paquete de ondas. En su lugar, puedes pensar en los qubits aproximadamente como porciones discretizadas de espacio. Aquí es el volumen de la red, donde cada elemento (celda unitaria) comprende dos qubits. Los operadores fermiónicos que vimos en la diapositiva anterior describen la amplitud de la función de onda en un sitio de red particular. Así que nuestro hamiltoniano contiene estos operadores fermiónicos de creación y destrucción. Por lo tanto, usamos la transformación de Jordan-Wigner para mapear estos operadores a los operadores de Pauli.
donde es el operador de Pauli y es . Una vez que tenemos un hamiltoniano en este formato, la parte difícil de la fase de mapeo ha terminado, y ahora puede escribirse fácilmente en un circuito en operadores de Pauli.
Conclusión
Hemos discutido cuatro ejemplos de cómo se han utilizado técnicas de mapeo específicas recientemente en el campo de la computación cuántica, desde las más sencillas hasta la aplicación de la transformación de Jordan-Wigner a la dinámica de hadrones. Gran parte de este material fue muy técnico, y si no lo has visto antes, puede resultar muy intimidante. Pero se vuelve más fácil cuanto más tiempo pases practicando -- por eso este curso se llama "Quantum Computing in Practice". Esto no es algo que alguien pueda simplemente tomar y salir corriendo desde el principio -- requiere algo de estudio, algo de rascarse la cabeza y probablemente algunos momentos frustrantes. Pero te animo a permanecer en esa incomodidad y explorar realmente las preguntas que surgen a medida que avanzas. Es la única forma de aprender.