Mejora de la estimación de energía de un Hamiltoniano de química con SQD
En este tutorial implementamos un patrón de Qiskit que muestra cómo post-procesar muestras cuánticas ruidosas para encontrar una aproximación al estado fundamental de un Hamiltoniano de química: la molécula en equilibrio en la base 6-31G. Seguiremos un enfoque de diagonalización cuántica basada en muestras para procesar muestras tomadas de un ansatz de circuito cuántico de 36 qubits (en este caso, un circuito LUCJ). Para tener en cuenta el efecto del ruido cuántico, se utiliza la técnica de recuperación de configuración.
El patrón se puede describir en cuatro pasos:
- Paso 1: Mapear al problema cuántico
- Generar un ansatz para estimar el estado fundamental
- Paso 2: Optimizar el problema
- Transpilar el ansatz para el Backend
- Paso 3: Ejecutar experimentos
- Extraer muestras del ansatz usando el primitivo
Sampler
- Extraer muestras del ansatz usando el primitivo
- Paso 4: Post-procesar resultados
- Bucle de recuperación de configuración auto-consistente
- Post-procesar el conjunto completo de muestras de cadenas de bits, usando conocimiento previo del número de partículas y la ocupación orbital promedio calculada en la iteración más reciente.
- Crear probabilísticamente lotes de submuestras a partir de las cadenas de bits recuperadas.
- Proyectar y diagonalizar el Hamiltoniano molecular sobre cada subespacio muestreado.
- Guardar la energía mínima del estado fundamental encontrada en todos los lotes y actualizar la ocupación orbital promedio.
- Bucle de recuperación de configuración auto-consistente
Para este ejemplo, el Hamiltoniano de electrones en interacción toma la forma genérica:
/ son los operadores fermiónicos de creación/aniquilación asociados al -ésimo elemento del conjunto de base y al espín . y son las integrales electrónicas de uno y dos cuerpos.
El flujo de trabajo de SQD con recuperación de configuración auto-consistente se muestra en el siguiente diagrama.
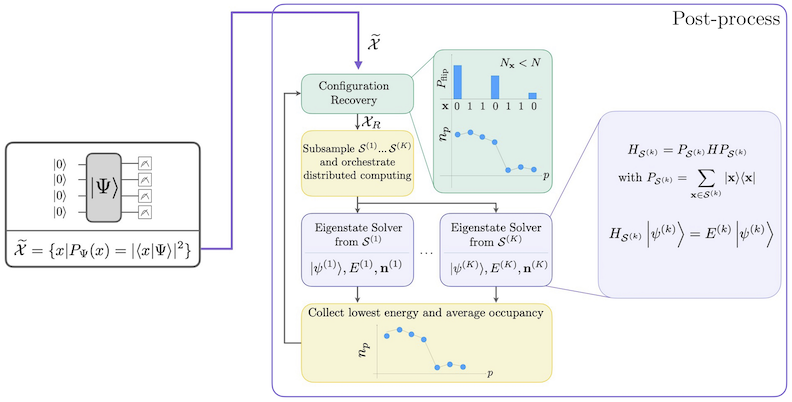
Se sabe que SQD funciona bien cuando el eigenestado objetivo es disperso: la función de onda está soportada en un conjunto de estados de base cuyo tamaño no crece exponencialmente con el tamaño del problema. En este escenario, la diagonalización del Hamiltoniano proyectado en el subespacio definido por :
produce una buena aproximación al eigenestado objetivo. El papel del dispositivo cuántico es producir muestras de los miembros de únicamente. Primero, un circuito cuántico prepara el estado en el dispositivo cuántico. Se utiliza la codificación de Jordan-Wigner. En consecuencia, los miembros de la base computacional representan estados de Fock (configuraciones/determinantes electrónicos). El circuito se muestrea en la base computacional, produciendo el conjunto de configuraciones ruidosas . Las configuraciones están representadas por cadenas de bits. El conjunto se pasa luego al bloque de post-procesamiento clásico, donde se utiliza la técnica de recuperación de configuración auto-consistente. En el marco de SQD, el papel del dispositivo cuántico es producir una distribución de probabilidad.
Paso 1: Mapear el problema a un circuito cuántico
En este tutorial, aproximaremos la energía del estado fundamental de una molécula de . Primero, especificaremos la molécula y sus propiedades. Luego, crearemos un ansatz local unitary cluster Jastrow (LUCJ) (circuito cuántico) para generar muestras desde una computadora cuántica para la estimación de la energía del estado fundamental.
Primero, especificaremos la molécula y sus propiedades.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
A continuación, crearemos el ansatz. El ansatz LUCJ es un circuito cuántico parametrizado, y lo inicializaremos con amplitudes t2 y t1 obtenidas de un cálculo CCSD.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
Usaremos el paquete ffsim para crear e inicializar el ansatz con las amplitudes t2 y t1 calculadas anteriormente. Dado que nuestra molécula tiene un estado de Hartree-Fock de capa cerrada, usaremos la variante de spin balanceado del ansatz UCJ, UCJOpSpinBalanced.
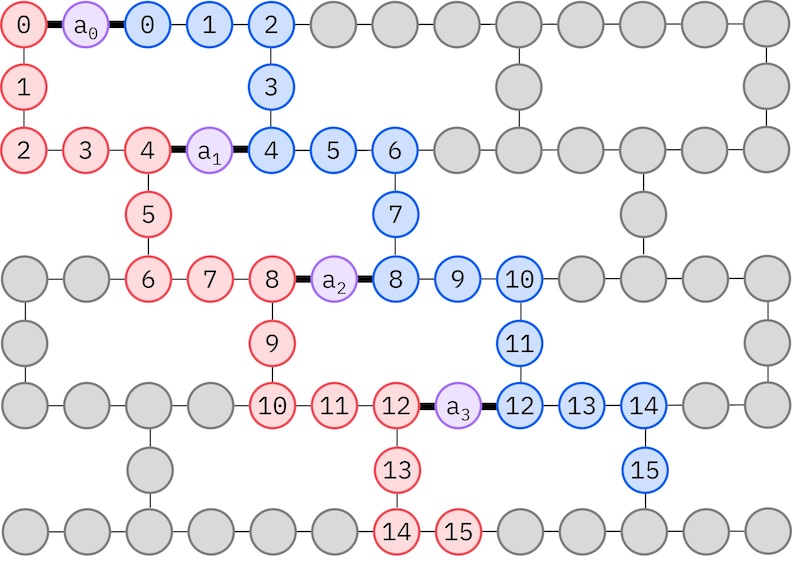
Como nuestro hardware IBM objetivo tiene una topología heavy-hex, adoptaremos el patrón zig-zag para las interacciones de qubits. En este patrón, los orbitales (representados por qubits) con el mismo espín están conectados con una topología lineal (círculos rojos y azules) donde cada línea toma una forma en zig-zag debido a la conectividad heavy-hex del hardware objetivo. De nuevo, debido a la topología heavy-hex, los orbitales de distintos espines tienen conexiones entre cada 4.º orbital (0, 4, 8, etc.) (círculos morados).
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Paso 2: Optimizar el problema
A continuación, optimizaremos nuestro circuito para un hardware objetivo. Necesitamos elegir el dispositivo de hardware a usar antes de optimizar nuestro circuito. Usaremos un Backend falso de 127 qubits de qiskit_ibm_runtime para emular un dispositivo real. Para ejecutar en una QPU real, simplemente reemplaza el Backend falso por uno real. Consulta la documentación de Qiskit IBM Runtime para más información.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
A continuación, recomendamos los siguientes pasos para optimizar el ansatz y hacerlo compatible con el hardware.
- Seleccionar qubits físicos (
initial_layout) del hardware objetivo que sigan el patrón zig-zag descrito anteriormente. Disponer los qubits en este patrón conduce a un circuito eficiente compatible con el hardware y con menos compuertas. - Generar un gestor de pases por etapas usando la función generate_preset_pass_manager de Qiskit con tu elección de
backendeinitial_layout. - Establecer la etapa
pre_initde tu gestor de pases por etapas enffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITincluye pases del Transpiler de Qiskit que descomponen las compuertas en rotaciones orbitales y luego las combinan, resultando en menos compuertas en el circuito final. - Ejecutar el gestor de pases en tu circuito.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
Paso 3: Ejecutar experimentos
Tras optimizar el circuito para su ejecución en el hardware, estamos listos para ejecutarlo en el hardware objetivo y recopilar muestras para la estimación de la energía del estado fundamental. Como solo tenemos un circuito, usaremos el modo de ejecución de trabajo (Job) de Qiskit Runtime y ejecutaremos nuestro circuito.
Nota: Hemos comentado el código para ejecutar el circuito en una QPU y lo dejamos como referencia para el usuario. En lugar de ejecutar en hardware real en esta guía, simplemente generaremos muestras aleatorias extraídas de la distribución uniforme.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
Paso 4: Post-procesar resultados
Ahora, ejecutamos el algoritmo SQD usando la función diagonalize_fermionic_hamiltonian. Consulta la documentación de la API para obtener explicaciones de los argumentos de esta función.
El solver incluido en el complemento SQD usa la implementación de CI seleccionado de PySCF, específicamente pyscf.fci.selected_ci.kernel_fixed_space. El ejemplo a continuación también muestra cómo pasar argumentos de palabras clave a esa función a través del solver incluido. Aquí pasamos el argumento max_cycle.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
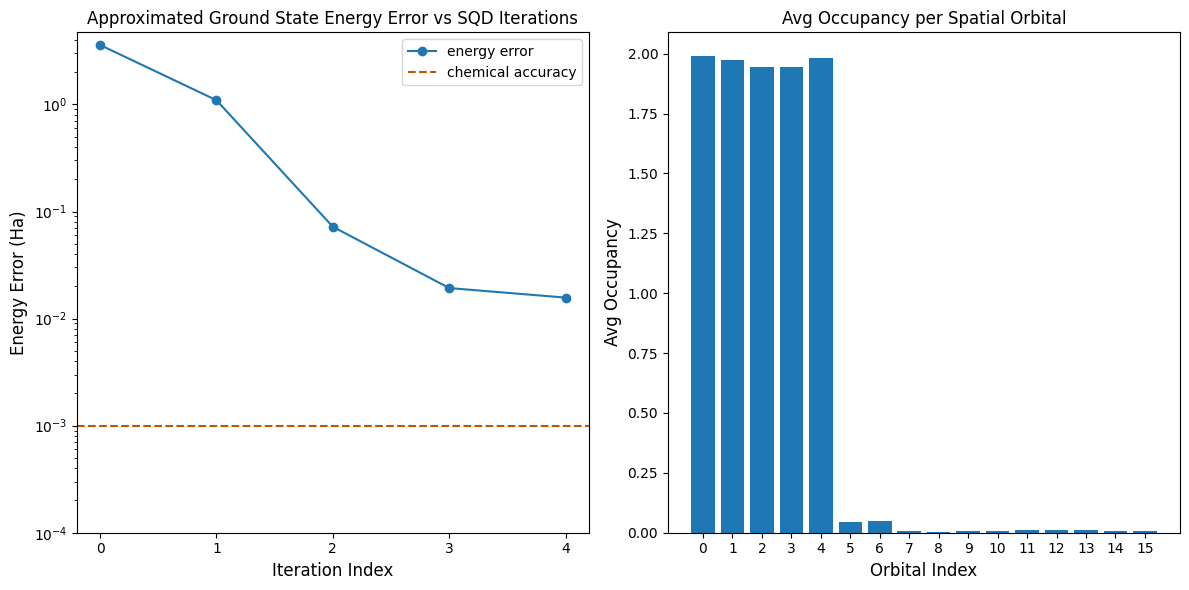
Ahora, graficamos los resultados.
La primera gráfica muestra que después de algunas iteraciones estimamos la energía del estado fundamental con una precisión de aproximadamente ~16 mH (la precisión química se acepta típicamente como 1 kcal/mol 1.6 mH). Recuerda que las muestras cuánticas en esta demostración eran ruido puro. La señal aquí proviene del conocimiento a priori de la estructura electrónica y el Hamiltoniano molecular.
La segunda gráfica muestra la ocupación promedio de cada orbital espacial después de la iteración final. Podemos ver que tanto los electrones de espín arriba como los de espín abajo ocupan los primeros cinco orbitales con alta probabilidad en nuestras soluciones.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha