Eigensolver cuántico variacional (VQE)
Para este módulo, los estudiantes deben tener un entorno de Python en funcionamiento y las últimas versiones de los siguientes paquetes instalados:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
Para configurar e instalar estos paquetes, consulta la guía Instalar Qiskit. Para ejecutar trabajos en computadoras cuánticas reales, los estudiantes deberán configurar una cuenta de IBM Cloud siguiendo los pasos de la guía Configura tu cuenta de IBM Cloud.
Este módulo ha sido probado y utilizó aproximadamente 8 minutos de tiempo de QPU. Esto es una estimación; tu uso real puede variar.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
Introducción
Desde el desarrollo del modelo mecánico cuántico a principios del siglo XX, los científicos han entendido que los electrones no siguen trayectorias fijas alrededor del núcleo de un átomo, sino que existen en regiones de probabilidad llamadas orbitales. Estos orbitales corresponden a niveles de energía específicos y discretos que los electrones pueden ocupar. Los electrones residen naturalmente en los niveles de energía más bajos disponibles, conocidos como el estado fundamental. Sin embargo, si un electrón absorbe suficiente energía, puede saltar a un nivel de energía superior y entrar en un estado excitado. Este estado excitado es temporal, y el electrón eventualmente regresará a un nivel de energía inferior, liberando la energía absorbida, con frecuencia en forma de luz. Este proceso fundamental de absorción y emisión de energía es importante para comprender cómo los átomos interactúan y forman enlaces.
Cuando los átomos se unen para formar moléculas, sus orbitales atómicos se combinan para formar orbitales moleculares. La disposición y los niveles de energía de los electrones dentro de estos orbitales moleculares dictan las propiedades de la molécula resultante y la fortaleza de los enlaces químicos. Por ejemplo, en la formación de una molécula de hidrógeno () a partir de dos átomos de hidrógeno individuales, el electrón de cada átomo ocupa orbitales atómicos. A medida que los átomos se acercan, estos orbitales atómicos se superponen y se combinan para formar nuevos orbitales moleculares — uno con menor energía (orbital enlazante) y otro con mayor energía (orbital antienlazante). Los dos electrones, uno de cada átomo de hidrógeno, ocuparán preferentemente el orbital enlazante de menor energía, lo que lleva a la formación de un enlace covalente estable que mantiene unida a la molécula . La diferencia de energía entre los átomos separados y la molécula formada, en particular la energía de los electrones en los orbitales moleculares, determina la estabilidad y las propiedades del enlace.
En las siguientes secciones, exploraremos este proceso de formación molecular, centrándonos en la molécula . Usaremos una computadora cuántica real, combinada con técnicas de optimización clásica, para encontrar la energía de este proceso simple pero fundamental. Este experimento proporcionará una demostración práctica de cómo la computación cuántica puede aplicarse para resolver problemas en química computacional, aportando perspectivas sobre el papel de la energía electrónica.
VQE: un algoritmo cuántico variacional para problemas de valores propios
Técnicas de aproximación en química: el principio variacional y el conjunto de base
Las contribuciones de Erwin Schrödinger a la mecánica cuántica no se limitan a introducir un nuevo modelo electrónico; fundamentalmente, estableció la mecánica ondulatoria desarrollando la famosa ecuación de Schrödinger dependiente del tiempo:
Aquí, es el operador hamiltoniano, que representa la energía total del sistema, y es la función de onda que contiene toda la información sobre el estado cuántico del sistema. (Nota: es la derivada total respecto al tiempo, y no incluimos explícitamente el valor propio de energía aquí.)
Sin embargo, en muchas aplicaciones prácticas — como determinar los niveles de energía permitidos de átomos y moléculas — usamos en cambio la ecuación de Schrödinger independiente del tiempo (ecuación de valores propios de energía), que se deriva de la forma dependiente del tiempo al asumir un estado estacionario. Un estado estacionario es un estado cuántico en el que la densidad de probabilidad de encontrar una partícula en un punto dado del espacio no cambia con el tiempo.
En esta forma, representa el valor propio de energía correspondiente al estado cuántico . El hamiltoniano incluye varias contribuciones energéticas, como la energía cinética de los electrones y los núcleos, las fuerzas atractivas entre electrones y núcleos, y las fuerzas repulsivas entre electrones.
Resolver la ecuación de valores propios de energía nos permite calcular los niveles de energía cuantizados de sistemas atómicos y moleculares. Sin embargo, para las moléculas, resolverla de forma exacta es difícil porque la función de onda , que describe la distribución espacial de los electrones, es compleja y de alta dimensionalidad.
Por ello, los científicos utilizan técnicas de aproximación para obtener soluciones prácticas y precisas. En este trabajo nos centraremos en dos métodos clave:
-
Principio variacional
Este método aproxima la función de onda y la ajusta para acercarse lo más posible a la energía objetivo, generalmente la energía del estado fundamental del sistema. La idea clave detrás del principio variacional es simple:
- Si proponemos una función de onda (una "función de prueba"), la energía calculada a partir de ella siempre será igual o mayor que la energía del estado fundamental () del sistema.
- Ajustando los parámetros en la función de prueba, , podemos obtener una aproximación cada vez mejor de la energía del estado fundamental.
- Su precisión depende en gran medida de la elección de la función de onda de prueba . Una función de prueba mal elegida puede llevar a una estimación de energía que diste mucho de ser precisa.
-
Aproximación del conjunto de base
El segundo método de aproximación aparece en la etapa de construcción de la función de onda: el enfoque del conjunto de base. En química cuántica, resolver la ecuación de Schrödinger de forma exacta para moléculas es prácticamente imposible. En cambio, aproximamos la función de onda multielectrónica compleja construyéndola a partir de funciones matemáticas más simples y predefinidas. Un conjunto de base es esencialmente una colección de estas funciones matemáticas conocidas, generalmente centradas en los átomos de la molécula, que se usan como bloques de construcción para representar la forma y el comportamiento de los electrones en el sistema. Piensa en ello como intentar recrear una escultura detallada usando solo un conjunto de piezas estándar de LEGO: cuantos más tipos y tamaños de piezas tengas (cuanto mayor sea el conjunto de base), más fielmente podrás aproximar la forma original.
Estas funciones base suelen estar inspiradas en las soluciones analíticas de sistemas simples como el átomo de hidrógeno, tomando formas como funciones gaussianas o de tipo Slater, aunque siguen siendo aproximaciones. En lugar de trabajar con los orbitales moleculares completos teóricamente "exactos" pero intratables, los expresamos como una combinación lineal (una suma con coeficientes) de estas funciones base. Este método se conoce como el enfoque de Combinación Lineal de Orbitales Atómicos (LCAO) cuando las funciones base se asemejan a orbitales atómicos. Optimizando los coeficientes en esta combinación lineal, podemos encontrar la mejor función de onda aproximada y la energía dentro de las limitaciones del conjunto de base elegido.
- Cuantas más funciones se incluyan en el conjunto de base, mejor será la aproximación, pero esto tiene el costo de un mayor esfuerzo computacional.
- Un conjunto de base pequeño proporciona una estimación aproximada, mientras que uno grande da resultados más precisos a expensas de requerir más recursos computacionales.
En resumen, para hacer los cálculos viables y reducir el costo computacional, usamos el principio variacional aproximando la función de onda, lo que reduce la complejidad computacional y permite una optimización iterativa para minimizar la energía. Mientras tanto, el enfoque del conjunto de base simplifica los cálculos representando los orbitales atómicos como una combinación de funciones predefinidas, en lugar de resolver directamente una función de onda continua.
Comprueba tu comprensión
Considera la función de onda de prueba , donde es una constante de normalización y es un parámetro ajustable.
(a) Normaliza la función de onda de prueba determinando de forma que .
Respuesta
Para normalizar la función de onda de prueba dada:
Usa la integral gaussiana:
establece y obtén:
(b) Calcula el valor esperado del hamiltoniano dado por donde , que corresponde a un potencial de oscilador armónico simple.
Respuesta
El hamiltoniano para un oscilador armónico es:
Valor esperado de la energía cinética
Tomando la segunda derivada:
Por lo tanto:
Usando los resultados estándar de integrales gaussianas:
Valor esperado de la energía potencial
Usando:
obtenemos:
Valor esperado de la energía total
(c) Usa el principio variacional para encontrar el óptimo minimizando .
Respuesta
Optimiza para la energía mínima
Diferencia
Resolviendo:
Sustituyendo en :
que coincide con la energía exacta del estado fundamental del oscilador armónico cuántico.
VQE (Eigensolver cuántico variacional)
El eigensolver cuántico variacional (VQE) es el método principal que usaremos para explorar el proceso , y aquí veremos qué es el VQE y cómo funciona. Pero primero hagamos una pausa y observemos algo muy importante mediante la siguiente pregunta de verificación.
Comprueba tu comprensión
Si ya tenemos tantas estrategias para los problemas de química, ¿por qué necesitamos una computadora cuántica? ¿Y cuál es el propósito de usar computadoras cuánticas y clásicas juntas?
Respuesta
La computación cuántica tiene la oportunidad de revolucionar la química al abordar problemas con los que las computadoras clásicas tienen dificultades debido al escalado exponencial de los estados cuánticos. Richard Feynman señaló célebremente que para simular la naturaleza, los cálculos también deben ser cuánticos [ref 1].
Por ejemplo, simular la cafeína con el conjunto de base más simple (STO-3G) requeriría bits, mucho más que el número total de estrellas en el universo observable () [ref 2]. Una computadora cuántica puede describir los orbitales electrónicos de la cafeína con 160 qubits.
Las computadoras cuánticas procesan de forma natural las interacciones cuánticas usando superposición y entrelazamiento, lo que proporciona una forma prometedora de habilitar simulaciones moleculares precisas. Además, podemos combinar las ventajas de las computadoras cuánticas (simulación electrónica) y las clásicas (preprocesamiento/posprocesamiento de datos, gestión del proceso algorítmico, optimización, etc.). Se espera que esto mejore el descubrimiento de materiales, el diseño de fármacos y las predicciones de reacciones, reduciendo los costosos experimentos de ensayo y error. [ref 3][ref 4]
Si quieres saber por qué se necesitan computadoras cuánticas para los problemas de química y por qué usar tanto recursos cuánticos como clásicos, consulta los siguientes artículos:
Ahora volvamos al VQE.
El VQE combina el poder de las computadoras cuánticas con las clásicas, usando fundamentalmente los principios variacionales para obtener la energía del estado fundamental del sistema. Para entender el VQE, primero divídelo en tres partes:
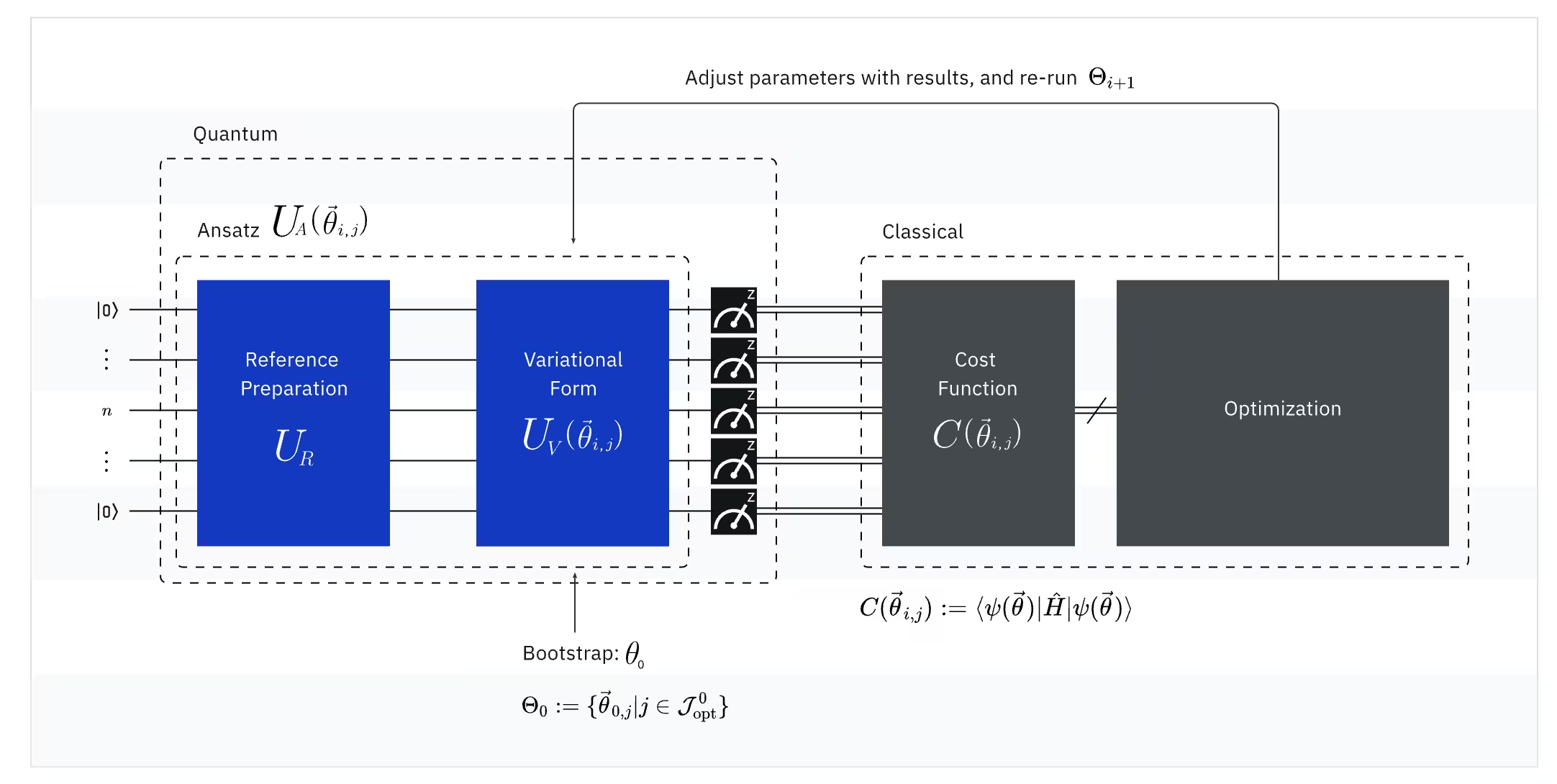
(Cuántico) Observable: el hamiltoniano molecular (energía de una molécula)
En el VQE, el hamiltoniano molecular/atómico es un observable, lo que significa que podemos medir su valor a través de un experimento. Nuestro objetivo es encontrar la energía más baja posible (la energía del estado fundamental) de la molécula. Para ello, usamos un estado cuántico de prueba, generado por un circuito cuántico parametrizado (ansatz). Medimos el observable y optimizamos el estado cuántico hasta alcanzar la energía más baja posible.
El conjunto de base utilizado para el hamiltoniano molecular determina el número de qubits requeridos y afecta directamente la precisión del VQE. Elegir el conjunto de base correcto es fundamental para equilibrar eficiencia y precisión. Para simplificar los cálculos sin cambiar el conjunto de base, podemos usar estrategias como imponer simetría y reducción del espacio activo. Muchas moléculas tienen formas simétricas (como una mariposa o un copo de nieve), lo que significa que algunas partes se comportan de la misma manera. En lugar de calcular todo por separado, podemos centrarnos solo en las partes únicas, ahorrando recursos cuánticos y aprovechando así la simetría. En la reducción del espacio activo, consideramos solo los orbitales importantes, ya que no todos los electrones impactan significativamente en la energía molecular. Los electrones cercanos al núcleo permanecen mayormente sin cambios, mientras que otros influyen en el enlace. Al aplicar estos métodos, podemos hacer el VQE más eficiente manteniendo la precisión.
Una vez que obtenemos un hamiltoniano molecular usando el conjunto de base apropiado y las estrategias anteriores, necesitamos transformar este hamiltoniano en uno adecuado para computadoras cuánticas. Mapear problemas a operadores de Pauli puede ser bastante complicado. Esto es especialmente cierto en química cuántica, que trabaja con partículas indistinguibles (electrones), ya que los qubits son distinguibles. No entraremos en los detalles de los mapeos aquí, pero te referimos a los siguientes recursos. Una discusión general sobre el mapeo de un problema a operadores cuánticos se puede encontrar en Quantum computing in practice. Una discusión más detallada sobre el mapeo de problemas de química en operadores cuánticos se puede encontrar en Quantum chemistry with VQE.
Para este módulo, te proporcionaremos los hamiltonianos (de un qubit) apropiados para y para que podamos centrarnos en usar la computadora cuántica. Estos hamiltonianos de un qubit se preparan usando el conjunto de base STO-6G y el mapeo de Jordan-Wigner, que es el mapeo más directo con la interpretación física más simple, porque mapea la ocupación de un spin-orbital a la ocupación de un qubit. Además, usamos una técnica de reducción de qubits mediante una simetría del hamiltoniano, que usa los patrones de cómo se comportan las ocupaciones de espín para reducir el número de qubits. Para la molécula , asumimos que la distancia entre los dos átomos de hidrógeno es 0.735 .
(Cuántico) Ansatz: la función de onda de prueba (cómo construir un estado cuántico trivial con un circuito cuántico)
Para el VQE, el ansatz (plural: ansätze) consta de dos componentes clave. El primero es la preparación del estado inicial, que configura el estado del qubit aplicando puertas cuánticas sin parámetros variacionales. El segundo componente es el circuito cuántico parametrizado, un circuito cuántico especial con parámetros ajustables, similar a los diales de una radio. Estos parámetros serán usados en la última parte — el optimizador clásico — para ayudarnos a alcanzar el mejor estado fundamental posible.
En la sección del principio variacional, aprendimos que la calidad del estado de prueba afecta la calidad de los resultados del algoritmo variacional. Esto significa que elegir un buen ansatz es importante en el VQE. Una vez más, este es un tema rico y complejo. No cubriremos aquí los diferentes tipos de ansatz ni sus orígenes. Si te interesa aprender más sobre circuitos cuánticos parametrizados y el ansatz, puedes explorar la lección Ansatz and variational form del curso de diseño de algoritmos variacionales, que proporciona explicaciones detalladas y ejemplos de ansätze.
Dado que vamos a usar un hamiltoniano de un qubit en este módulo, necesitamos un circuito cuántico parametrizado de un qubit como ansatz. Veremos tres tipos de ansätze de un qubit en la siguiente sección. Los compararemos y discutiremos consideraciones clave para seleccionar un ansatz.
(Clásico) Optimizador: ajuste fino del circuito cuántico
Una vez que la computadora cuántica mide la energía del observable a partir del ansatz, los parámetros del ansatz y el valor de energía se envían al optimizador clásico para su ajuste. Este proceso de optimización se realiza en una computadora clásica, generalmente usando paquetes científicos de propósito general como SciPy.
El optimizador clásico trata la energía medida como una función de costo. En los problemas de optimización, una función de costo (también llamada a veces función objetivo) es una función matemática que mide qué tan "buena" es una solución particular. El objetivo del optimizador es encontrar el conjunto de parámetros que minimice esta función de costo. En el contexto de encontrar la energía del estado fundamental de una molécula, la propia energía sirve como función de costo — queremos encontrar los parámetros para nuestro circuito cuántico (nuestra "solución") que produzcan la energía más baja posible. El optimizador clásico usa este valor de energía medido (el costo) y determina el siguiente conjunto de parámetros optimizados para el ansatz cuántico. Estos parámetros actualizados se envían de vuelta al circuito cuántico, y el proceso se repite. Con cada iteración, el optimizador clásico ajusta los parámetros para intentar reducir la energía (minimizar la función de costo) hasta que se cumple un criterio de convergencia predefinido, asegurando idealmente que se encuentre la energía más baja posible (correspondiente al estado fundamental de la molécula para esa distancia de enlace y conjunto de base).
Existen muchas estrategias de optimización proporcionadas por paquetes científicos como SciPy. Puedes encontrar más en la lección Optimization loops del curso Variational algorithm design. Aquí usaremos COBYLA (Optimización con restricciones por aproximaciones lineales), un algoritmo de optimización adecuado para paisajes de energía complicados. En particular, COBYLA no intenta calcular un gradiente de la función estudiada; esto se denomina optimizador libre de gradiente. Imagina que intentas encontrar el pico más alto en una cordillera con los ojos cerrados. Como no puedes ver todo el paisaje, das pequeños pasos en diferentes direcciones, verificando si estás subiendo o bajando. COBYLA funciona de manera similar: se mueve por el espacio de parámetros, probando diferentes valores, mejorando gradualmente el resultado hasta encontrar el mejor.
Ahora estás listo para llevar a cabo un cálculo de VQE. Para ello, prueba la siguiente pregunta de verificación, que resume el proceso general.
Comprueba tu comprensión
Completa los espacios en blanco con los términos correctos para completar el resumen del proceso de VQE y luego haz clic para ver tus respuestas.
El VQE es un algoritmo cuántico variacional que combina el poder de (1) ________ y la computación clásica, usado para encontrar la (2) __________ de una molécula. El proceso comienza definiendo el (3) __________, que representa la energía total del sistema y actúa como el observable en las mediciones cuánticas. A continuación, preparamos un (4) __________, un circuito cuántico con parámetros ajustables que representa la función de onda de prueba de la molécula. Estos parámetros se optimizan usando un (5) __________, un algoritmo clásico que ajusta los parámetros iterativamente para minimizar la energía medida. En la discusión anterior usamos el optimizador (6) __________, que refina los parámetros del ansatz sin necesitar cálculos de derivadas. El proceso continúa hasta que alcanzamos la (7) __________, lo que significa que hemos encontrado la energía más baja posible de la molécula.
Banco de palabras:
- optimizador clásico
- energía del estado fundamental
- eficiente en hardware
- ansatz
- hamiltoniano molecular
- COBYLA
- computación cuántica
- convergencia
Respuesta
1 → computación cuántica
2 → energía del estado fundamental
3 → hamiltoniano molecular
4 → ansatz
5 → optimizador clásico
6 → COBYLA
7 → convergencia
Calcula la energía del estado fundamental de un átomo de hidrógeno con VQE
Ahora vamos a usar lo que hemos aprendido para calcular la energía del estado fundamental de un átomo de hidrógeno. A lo largo del módulo, usaremos un marco de trabajo para computación cuántica conocido como "patrones de Qiskit", que divide los flujos de trabajo en los siguientes pasos:
- Paso 1: Mapear las entradas clásicas a un problema cuántico
- Paso 2: Optimizar el problema para la ejecución cuántica
- Paso 3: Ejecutar usando las primitivas de Qiskit Runtime
- Paso 4: Posprocesamiento y análisis clásico

En general, seguiremos estos pasos.
Empecemos cargando algunos paquetes necesarios, incluyendo las primitivas de Qiskit Runtime. También seleccionaremos el computador cuántico menos ocupado que tengamos disponible.
A continuación hay código para guardar tus credenciales en el primer uso. Asegúrate de eliminar esta información del notebook después de guardarla en tu entorno, para que tus credenciales no se compartan accidentalmente cuando compartas el notebook. Consulta Configura tu cuenta de IBM Cloud e Inicializa el servicio en un entorno no confiable para obtener más orientación.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
La celda a continuación te permitirá cambiar entre el simulador o hardware real en cualquier momento del notebook. Te recomendamos ejecutarla ahora:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
Paso 1: Mapear el problema a circuitos cuánticos y operadores
Comenzamos nuestro cálculo de VQE definiendo el Hamiltoniano para la molécula de hidrógeno () a una distancia de enlace específica. Este Hamiltoniano representa la energía total del sistema en términos de operadores de qubits, habiendo sido producido y mapeado desde el sistema molecular mediante un procedimiento estándar: 1) empleando el conjunto de bases STO-6G (una colección específica de funciones matemáticas usadas para aproximar los orbitales electrónicos), 2) aplicando el mapeo de Jordan-Wigner (una técnica para traducir operadores fermiónicos que describen electrones en operadores de qubits), y 3) realizando una reducción de qubits usando la paridad del Hamiltoniano para simplificar el problema.
Como explicamos anteriormente, las energías del estado fundamental calculadas dependen en gran medida de la selección del conjunto de bases y de la geometría molecular (como la distancia de enlace). Para esta configuración específica y después de estas transformaciones, el Hamiltoniano de qubits resultante es sencillo:
Aquí, representa el operador identidad y representa el operador de Pauli-Z, que actúa sobre un único qubit. Los coeficientes se derivan de las integrales calculadas usando el conjunto de bases STO-6G en esta distancia de enlace particular con la transformación adecuada.
Con este Hamiltoniano definido, ahora podemos usar VQE para calcular su energía del estado fundamental. Es útil comparar nuestra energía calculada del estado fundamental con valores esperados. Para un átomo de hidrógeno (H) único y aislado, la energía del estado fundamental es exactamente -0.5 Hartree (en ausencia de efectos relativistas). Calculemos la energía exacta del estado fundamental de nuestro Hamiltoniano de qubits específico tal como se definió arriba y comparémosla con valores conocidos relevantes.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
A continuación, necesitamos un circuito cuántico parametrizado, un ansatz, para preparar una función de onda de prueba para el estado fundamental. El objetivo es encontrar los parámetros que minimicen el valor esperado de energía . La elección del ansatz es crucial porque determina el conjunto de estados cuánticos posibles que nuestro circuito puede preparar. Un "buen" ansatz es aquel lo suficientemente flexible para representar un estado muy cercano al verdadero estado fundamental del Hamiltoniano que estamos estudiando, pero no tan complejo que requiera demasiados parámetros o un circuito demasiado profundo para los computadores cuánticos actuales.
Aquí probaremos tres ansätze de un qubit diferentes para ver cuál proporciona mejor "cobertura" de los posibles estados cuánticos en los que puede estar un único qubit. La "cobertura" se refiere al rango de estados cuánticos que el circuito ansatz puede producir al variar sus parámetros.
Usaremos tres ansätze basados en diferentes combinaciones de puertas de rotación de un solo qubit:
- Ansatz con una puerta de rotación en un eje: Este ansatz usa rotaciones alrededor de un único eje (). En la esfera de Bloch, esto corresponde a moverse únicamente a lo largo de un círculo específico. Es el menos flexible y cubre un conjunto limitado de estados.
- Dos ansätze con puertas de rotación en dos ejes: Estos ansätze combinan rotaciones alrededor de dos ejes diferentes ( y ). Esto nos permite alcanzar una porción mayor de la esfera de Bloch, en comparación con una rotación de un solo eje.
Al comparar los resultados de VQE obtenidos con estos tres ansätze, podemos ver cómo la flexibilidad y la cobertura del espacio de estados del ansatz afectan nuestra capacidad para encontrar la verdadera energía del estado fundamental de nuestro Hamiltoniano simplificado. Un ansatz más flexible tiene el potencial de encontrar una mejor aproximación, pero también puede ser más difícil de optimizar para el optimizador clásico.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
Ahora vamos a generar 5000 números aleatorios para cada parámetro y graficar la distribución de estados cuánticos aleatorios, generados por los tres ansätze con estos parámetros aleatorios. Puedes pensar en estos parámetros como rotaciones alrededor de diferentes ejes en una superficie esférica. Para ver la distribución de los estados cuánticos, usaremos la esfera de Bloch, una esfera tridimensional que muestra el estado de un único qubit. Cualquier punto en la esfera representa un posible estado del qubit, donde los polos norte y sur son como el "0" y el "1" clásicos, pero el qubit también puede estar en cualquier punto intermedio, mostrando propiedades cuánticas especiales como la superposición. Primero, prepara las funciones necesarias para graficar la esfera de Bloch 3D y genera 5000 parámetros aleatorios.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
Veamos cómo funciona nuestro primer ansatz.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
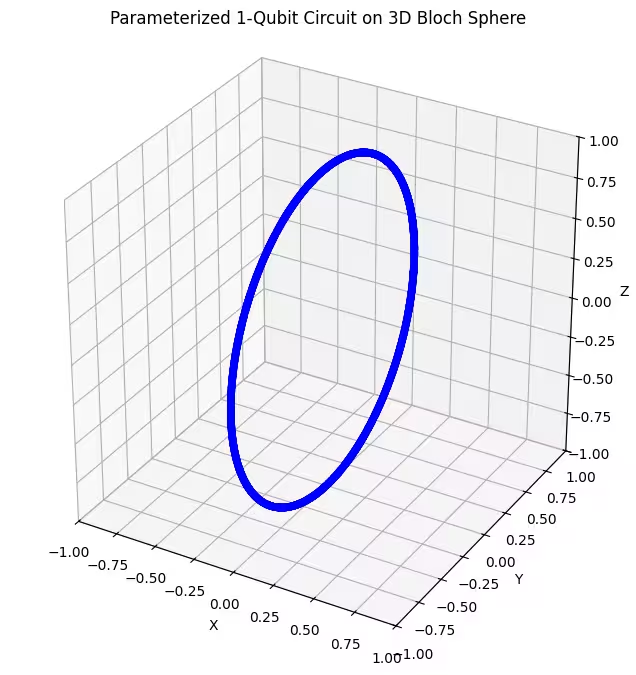
Podemos ver que nuestro primer ansatz devuelve estados cuánticos distribuidos en forma de anillo en la esfera de Bloch. Esto tiene sentido, porque solo le hemos dado al ansatz un único parámetro de rotación. Por lo tanto, solo puede producir estados rotados alrededor de un eje. Comenzando desde el punto y rotando alrededor de un eje siempre se obtiene un anillo. Ahora revisemos nuestro segundo ansatz, que tiene dos puertas de rotación ortogonales: Rx y Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
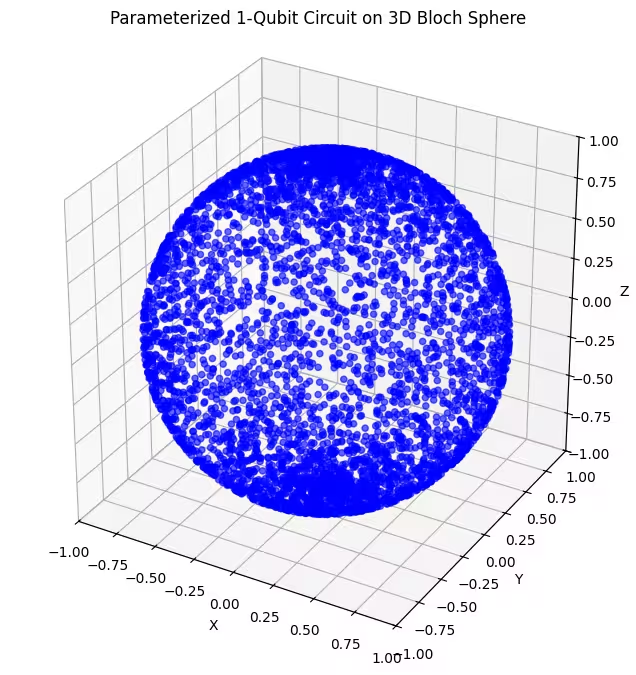
Aquí podemos ver que nuestro segundo ansatz cubre una porción mayor de la esfera de Bloch, aunque nótese que los puntos están más concentrados alrededor de los polos y más dispersos alrededor del ecuador. Ahora es el momento de revisar nuestro último ansatz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
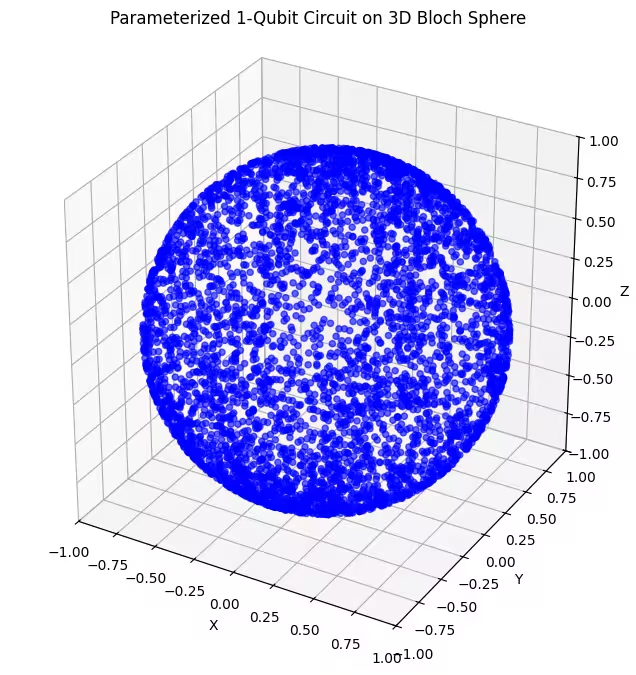
Aquí puedes ver estados cuánticos distribuidos de forma más uniforme, generados por nuestro último ansatz.
Como se menciónó, lo mejor es conocer el estado fundamental que se busca y usar un ansatz bien adaptado para explorar estados cercanos a ese estado fundamental. Por ejemplo, si supiéramos que nuestro estado fundamental está cerca de un polo, podríamos seleccionar el ansatz 2. Por simplicidad, nos quedaremos con el ansatz 3, que explora la esfera de Bloch completa de forma uniforme.
Ahora que hemos seleccionado nuestro ansatz, dibujemos el circuito.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

Paso 2: Optimizar para el hardware destino
Cuando ejecutamos un cálculo en un computador cuántico real, no solo nos importa la lógica del circuito cuántico. También nos importan cosas como qué operaciones puede realizar ese computador cuántico en particular, y dónde están ubicados los qubits que vamos a usar dentro del computador cuántico. ¿Están uno al lado del otro? ¿Están lejos? Por lo tanto, el siguiente paso es reescribir nuestro circuito usando puertas que sean naturales para el computador cuántico que vamos a usar, teniendo en cuenta la disposición de los qubits. Esto se puede hacer mediante transpilación: después de este proceso, puedes ver nuestro sencillo ansatz convertido en un conjunto diferente de puertas, y nuestros qubits abstractos serán mapeados a qubits físicos en un computador cuántico real.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

Puedes ver que las puertas rx, rz de nuestro ansatz fueron convertidas en una serie de puertas rz, sx, que son las puertas nativas de nuestro backend. Además, puedes ver que nuestro q0 ahora está mapeado al quinto qubit físico. También necesitamos mapear nuestro Hamiltoniano de acuerdo con estos cambios, como se muestra en el siguiente código:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
Paso 3: Ejecutar en el hardware destino
Ahora es el momento de ejecutar nuestro VQE en una QPU real. Para ello, primero necesitamos una función de costo para el proceso de optimización, que evalúe el valor esperado del Hamiltoniano con un estado cuántico generado por el ansatz. ¡No te preocupes! No necesitas programar todo por tu cuenta. Hemos preparado una función para esto, y todo lo que tienes que hacer es ejecutar la celda a continuación.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
Por último, preparamos los parámetros iniciales para nuestro ansatz y su proceso de optimización. Puedes usar simplemente todos ceros o valores aleatorios. Hemos seleccionado los parámetros iniciales a continuación, pero siéntete libre de comentar o descomentar las líneas de la celda para muestrear los parámetros de forma aleatoria y uniforme entre 0 y .
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
¡Felicitaciones! Acabas de completar tu primer experimento de química cuántica con éxito. Podemos ver una diferencia entre la energía exacta del estado fundamental del Hamiltoniano y la nuestra, pero como usamos una técnica de mitigación de errores predeterminada (que corrige errores de lectura), la diferencia es pequeña. ¡Es un muy buen comienzo!
Nota: Puedes obtener un mejor resultado estableciendo un nivel de mitigación de errores usando resilience_level. El valor predeterminado es 1, y si estableces un valor mayor, usará más tiempo de QPU pero podría devolver un mejor resultado.
Paso 4: Posprocesamiento
Es el momento de ver cómo funciónó nuestro optimizador clásico. Ejecuta la celda a continuación y observa el patrón de convergencia.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Comenzamos con un valor inicial bastante bueno, de modo que obtuvimos un buen valor final en solo 10 pasos. Puedes ver picos grandes y pequeños, y esta es la característica típica del optimizador COBYLA: explora el espacio como si no pudiera ver el paisaje y ajusta el tamaño de los pasos con cada medición.
Comprueba tu comprensión
¿Cuál es tu observación? ¿Qué parte del proceso anterior está abierta a mejoras para obtener resultados más cercanos a los valores teóricos, o más cercanos a la energía precisa del estado fundamental del Hamiltoniano? ¿Qué aspectos hay que tener en cuenta para esto?
Respuesta
Lo primero a considerar es el cambio en el conjunto de bases utilizado para calcular el Hamiltoniano de las moléculas. Como se menciónó anteriormente, la energía del estado fundamental del átomo de H es -0.5 Hartree, como es bien sabido, y la base STO-6G que hemos elegido no es suficiente para derivar este valor con precisión.
Elegir un tipo de base más compleja aumenta el número de qubits usados por el Hamiltoniano; por lo tanto, necesitamos seleccionar un ansatz más complejo y adecuado para problemas de química.
Lo siguiente a optimizar es la gestión del ruido en la QPU. Las técnicas de mitigación de errores más avanzadas producen mejores resultados, pero pueden tardar más en aplicarse. Considera también cómo el shot_number afecta los resultados.
Por último, también se puede lograr un mejor rendimiento de convergencia probando diferentes optimizadores.
Calcula la energía del estado fundamental de la molécula de hidrógeno con VQE
Ahora que hemos visto el proceso general de VQE usando átomos de , calcularemos la energía del estado fundamental de la molécula de forma más rápida.
Paso 1: Mapear el problema a circuitos cuánticos y operadores
Aquí también te proporcionamos un Hamiltoniano de un qubit que utiliza la base STO-6G y la transformación de Jordan-Wigner, con reducción de qubits mediante una simetría del Hamiltoniano. Ten en cuenta que usamos una distancia atómica de 0.735 entre los dos átomos de hidrógeno.
A diferencia del cálculo de un átomo de hidrógeno simple (), para calcular el estado fundamental de una molécula de hidrógeno (), también debemos considerar la fuerza repulsiva que actúa entre los núcleos de los dos átomos de hidrógeno, además de la energía asociada a los orbitales electrónicos. En este paso daremos este valor como una constante, y lo calcularemos realmente en el problema de verificación.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
Paso 2: Optimizar para el hardware de destino
Como el número de qubits que utiliza el VQE y el Hamiltoniano anteriores es el mismo que el del backend que se usará para la ejecución, utilizaremos el ansatz existente y su forma optimizada.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
Paso 3: Ejecutar en el hardware de destino
Ahora es momento de hacer los cálculos en la QPU real. Casi todo es igual, pero usaremos el punto inicial adecuado para ajustar el Hamiltoniano. Además, en la parte iterativa, algunos ajustes del Estimator, que se usa para calcular las expectativas del Hamiltoniano para el ansatz en la QPU, se configurarán de forma ligeramente diferente respecto a los cálculos anteriores. Hablaremos de este cambio con más detalle en una pregunta de verificación.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
Aunque VQE proporciona teóricamente una cota superior a la verdadera energía del estado fundamental, las implementaciones prácticas en hardware cuántico real o simulado con ruido, así como las aproximaciones realizadas al preparar el Hamiltoniano (como los conjuntos de base o la reducción de qubits), pueden introducir errores que a veces resultan en una energía medida ligeramente inferior al valor teórico exacto o a una referencia numérica específica. Aunque hay algunos errores, los resultados parecen satisfactorios, especialmente dado el pequeño número de pasos. Ahora, terminemos este cálculo de VQE viendo cómo funciónó el optimizador.
Paso 4: Post-procesamiento
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Comprueba tu comprensión
Calculemos la energía de repulsión nuclear de la molécula de , que incluimos como valor constante (0.71997 Hartree).

Usa la ley de Coulomb y las unidades atómicas para asegurarte de obtener el valor en Hartree.
Respuesta
Como ambos núcleos de hidrógeno tienen carga positiva, se repelen entre sí por fuerza electrostática. Esta repulsión se describe mediante la ley de Coulomb:
donde es la carga del protón, es la permitividad del vacío y es la distancia entre los dos núcleos, medida en metros o radios de Bohr en unidades de julios (J).
Para calcular esta energía en Hartrees, necesitamos convertir la ecuación anterior al sistema de Unidades Atómicas (UA). En UA, , y el radio de Bohr () es 1 y se convierte en la escala de longitud fundamental en UA. Con estas simplificaciones, la ley de Coulomb se reduce a:
,
donde debe medirse en radios de Bohr ().
Para convertir la separación nuclear dada en a , necesitamos la siguiente relación de conversión:
por lo que se convierte en .
Por lo tanto, la energía de repulsión nuclear del dado es
Calcula la energía de reacción de
¡Ahora pongamos en práctica lo que hemos obtenido! Has utilizado VQE, un solver cuántico variacional de valores propios, para calcular la energía del estado fundamental del átomo y de la molécula . Lo que queda es usar los valores calculados para obtener la energía de reacción del proceso .
La energía de reacción es el cambio de energía que ocurre cuando las sustancias reaccionan para formar nuevas sustancias. Imagina que estás construyendo algo: a veces necesitas aportarle energía (como apilar bloques) y otras veces se libera energía (como una pelota rodando cuesta abajo). En química, las reacciones absorben energía (endotérmicas) o la liberan (exotérmicas).
La energía de reacción del proceso se puede calcular con la siguiente fórmula:
Ejecutando la celda de abajo, veamos esto de forma visual. Aquí usaremos el valor exacto del estado fundamental de cada Hamiltoniano, y compararemos la energía de reacción de la solución exacta con los resultados de VQE.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

Como se muestra en la figura, aunque hay algunos errores, la energía exacta del estado fundamental de los Hamiltonianos y la energía de reacción calculada usando los resultados de VQE son similares, cercanas a -0.2 Hartree.
Cabe señalar aquí que la energía de reacción de este proceso tiene un valor negativo, lo que significa que se libera energía a través del proceso y que la molécula resultante tiene una energía menor que la de dos átomos individuales. 6. Conclusión
Resumamos lo que hemos aprendido hasta ahora.
Primero vimos dos importantes técnicas de aproximación necesarias para resolver problemas de química cuántica: el principio variacional y la elección del conjunto de base, ambas fundamentales para VQE. Exploramos el principio variacional a mano, calculando la energía del estado fundamental del oscilador armónico simple.
A continuación, exploramos VQE, un algoritmo ampliamente utilizado para calcular la energía del estado fundamental de un sistema cuántico. Ejecutamos código para calcular las energías del estado fundamental del hidrógeno atómico () y de la molécula de hidrógeno (). En particular, aprendimos que es necesario obtener el Hamiltoniano molecular adecuado para el sistema y transformarlo a una forma ejecutable en un computador cuántico. También vimos que el ansatz, un circuito cuántico parametrizado, es necesario para preparar estados cuánticos de prueba dentro de VQE, y discutimos la importancia de elegir una estructura de circuito ansatz apropiada. También aprendimos que VQE se basa en un proceso de optimización iterativo usando un computador clásico, que guía el circuito cuántico para encontrar el estado de menor energía, y vimos cómo converge el proceso.
Finalmente, usamos las energías del estado fundamental de y obtenidas mediante VQE para calcular la energía de reacción del proceso .
VQE es un potente algoritmo cuántico de corto plazo, pero es importante conocer sus limitaciones. El rendimiento de VQE depende en gran medida de la elección del ansatz: encontrar un ansatz que se pueda preparar eficientemente y que represente con precisión el verdadero estado fundamental se vuelve difícil para moléculas más grandes y complejas. Además, el hardware cuántico actual es susceptible al ruido, lo que puede afectar la precisión de los resultados de VQE, especialmente para circuitos más profundos o un mayor número de qubits. A pesar de estos desafíos, VQE sirve como algoritmo fundacional, y la investigación en curso está explorando métodos variacionales más sofisticados y técnicas de mitigación de errores para ampliar los límites de lo posible en química cuántica con computadores cuánticos de corto plazo. Por ejemplo, algoritmos como la Diagonalización Cuántica Basada en Muestras (SQD, por sus siglas en inglés) están siendo desarrollados, los cuales aprovechan muestras obtenidas de circuitos cuánticos combinadas con diagonalización clásica en un subespacio para mejorar la estimación de energía y abordar algunas de las limitaciones de VQE, particularmente en cuanto a eficiencia de medición y robustez frente al ruido.
Revisión y preguntas
Conceptos clave:
- El algoritmo cuántico variacional es un paradigma de computación en el que un computador clásico y un computador cuántico trabajan juntos para resolver un problema.
- En VQE, comenzamos con un Hamiltoniano de nuestro sistema y lo mapeamos a qubits para su ejecución en el computador cuántico. Seleccionamos un circuito cuántico parametrizado, un ansatz, y realizamos mediciones repetidas variando los parámetros del ansatz hasta alcanzar el valor de energía más bajo. La búsqueda en el espacio de parámetros se realiza mediante un optimizador clásico. Para obtener buenos resultados, es necesario seleccionar un buen ansatz y un optimizador apropiado.
- La energía de reacción es el cambio total de energía en una reacción química, determinado por la diferencia entre la energía de los reactivos y la de los productos.
Verdadero/falso
- El principio variacional establece que el valor esperado de la energía para cualquier función de onda de prueba es siempre mayor o igual que la verdadera energía del estado fundamental.
- Un conjunto de base es una colección de funciones utilizadas para aproximar funciones de onda cuánticas.
- VQE es un algoritmo cuántico usado para resolver exactamente la ecuación de Schrödinger para un Hamiltoniano dado.
- En VQE, se usa un circuito cuántico parametrizado (un ansatz) para preparar funciones de onda de prueba.
- La elección del optimizador en VQE (por ejemplo, COBYLA, SPSA o ADAM) no afecta la calidad del resultado.
- El
Estimatorde Qiskit se usa para calcular directamente los valores esperados de los Hamiltonianos en VQE.
Preguntas de opción múltiple:
- ¿Cuál es el propósito del Hamiltoniano en VQE?
- A) Generar estados cuánticos aleatorios
- B) Determinar la energía de los estados cuánticos
- C) Optimizar circuitos cuánticos
- D) Crear entrelazamiento
- ¿Cuál es el objetivo principal del algoritmo VQE?
- A) Encontrar la energía del estado fundamental de un Hamiltoniano
- B) Crear entrelazamiento entre qubits
- C) Realizar la búsqueda de Grover
- D) Romper el cifrado RSA
- ¿Cuántos estados cuánticos se generan en este notebook para comparar el ansatz?
- A) 100
- B) 1000
- C) 5000
- D) 10.000
- ¿Por qué se necesita un optimizador clásico en VQE?
- A) Para realizar mediciones cuánticas
- B) Actualizar los parámetros del ansatz para minimizar la energía
- C) Para entrelazar qubits
- D) Para generar aleatoriedad cuántica
- ¿Por qué el ansatz está diseñado para ser parametrizado?
- A) Para permitir la preparación de estados cuánticos
- B) Para permitir explorar un amplio espacio de estados cuánticos
- C) Para reducir la complejidad del circuito
- D) Para medir valores propios directamente
- ¿Cuál de las siguientes es la afirmación más correcta sobre cómo elegir un buen ansatz?
- A) Un ansatz debe producir estados distribuidos uniformemente sobre la esfera de Bloch, o fallará.
- B) Un ansatz debe adaptarse a tu sistema para asegurarse de que pueda generar estados cercanos al estado fundamental.
- C) Un ansatz debe producir estados aleatorios usando sus parámetros variacionales.
- D) Un mejor ansatz siempre tiene más parámetros variacionales.
(Opcional) Apéndice: Sobrecarga del optimizador según la complejidad del ansatz
VQE enfrenta varios desafíos bien conocidos [ref 6], y los siguientes están relacionados con lo que hemos aprendido anteriormente.
- Desafíos en la selección del ansatz
Existe un desafío inherente en la selección del ansatz variacional adecuado. Los ansätze inspirados en química (como UCCSD) proporcionan precisión física pero requieren circuitos profundos, mientras que los ansätze eficientes para hardware tienen circuitos más superficiales pero pueden carecer de interpretabilidad física. Además, muchos ansätze introducen parámetros variacionales excesivos que contribuyen poco a mejorar la precisión pero aumentan significativamente la dificultad de optimización.
- Dificultades de optimización
El paisaje de optimización de VQE puede tener regiones donde los gradientes se desvanecen exponencialmente (mesetas áridas o "barren plateaus"), lo que dificulta que los optimizadores clásicos actualicen los parámetros variacionales de forma eficiente. Para esto, los investigadores han intentado usar distintos tipos de optimizadores, tanto basados en gradientes como libres de gradientes, pero ambos enfrentan desafíos. Los optimizadores basados en gradientes sufren de mesetas áridas, mientras que los métodos libres de gradientes requieren un gran número de evaluaciones de la función.
- Sobrecarga del optimizador
Otro desafío bien conocido es la sobrecarga del optimizador, que está relacionada con la escala del problema. Los circuitos cuánticos requeridos para VQE crecen en profundidad y complejidad a medida que aumenta el tamaño del problema; esto también suele incrementar el número de parámetros a optimizar. El proceso de optimización se vuelve intratable a medida que aumenta el número de parámetros, lo que lleva a una convergencia lenta y dificultades para encontrar la solución óptima.
Aquí veremos estos desafíos usando VQE para una molécula de , con dos tipos diferentes de ansätze.
(Nota: Esto puede requerir más tiempo de QPU, así que no dudes en usar un simulador si no tienes suficiente tiempo.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
Ahora ejecutemos un VQE con un punto inicial de todos unos, con un máximo de 20 pasos, y comparemos la convergencia de ambas ejecuciones.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

La gráfica anterior demuestra claramente que el proceso de optimización del ansatz con más variables tarda más en alcanzar una convergencia estable.
En lugar de depender de circuitos simples de un solo qubit y un ansatz sencillo, la complejidad de la optimización aumenta cuando se requieren circuitos cuánticos más grandes y ansätze con estructuras más complejas. Esto pone de relieve un desafío bien conocido en VQE: la sobrecarga del optimizador.
Los investigadores continúan desarrollando diversas metodologías avanzadas que pueden usar computadores cuánticos para resolver problemas de química. Puedes acceder a una variedad de materiales educativos en IBM Quantum Learning.
Referencias
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)